de primaire structuur van een eiwit wordt gedefinieerd als de sequentie van aminozuren waaruit het is samengesteld. Deze opeenvolging bepaalt uiteindelijk de vorm die de proteã ne, volgens de ruimtelijke beperkingen op de regeling van de atomen in de proteã ne, de chemische eigenschappen van de residu ‘ s van het componentaminozuur, en het milieu van de proteã ne aanneemt.
de peptidebindingen die aminozuurresiduen in een polypeptide met elkaar verbinden, worden gevormd in een condensatiereactie tussen de zure carboxylgroep van één aminozuur en de basisaminogroep van een ander aminozuur. In de context van een peptide, wordt de amidegroep (CO–NH) bedoeld als de peptidegroep.
cruciaal voor een goed begrip van de eiwitstructuur is een kennis van de structuur van de peptidebinding. Linus Pauling, in de jaren 1930, gebruikte Röntgenstraal diffractie om de aard van de peptide band te onderzoeken die tussen twee aminozuren wordt gevormd. Hij meldde dat de peptidegroep (CO–NH) een stijve vlakke structuur heeft. Deze structuur is te wijten aan interacties tussen elektronen van de dubbele binding van de carbonylgroep en die van de C–N binding (Figuur 2), zodat deze laatste partiële (ongeveer 40%) dubbele binding eigenschappen verwerft.

dit effect is een voorbeeld van resonantie die kan worden gezien als het delen van elektronen tussen bindingen. Aangezien enkelvoudige bindingen tussen twee atomen langer zijn dan dubbele bindingen tussen dezelfde twee atomen, verschillen de lengtes van de C–N-en C=O-bindingen in de peptidegroep van deze bindingen in andere contexten waar resonantie niet voorkomt. Het partiële dubbele bindingskarakter van C–N in de peptidegroep betekent dus dat deze binding korter is dan voorspeld zou worden voor een enkele binding van C–N, terwijl de c=o-binding, die een gedeeltelijk enkel bindingskarakter heeft als gevolg van resonantie, langer is dan voorspeld zou worden voor een dubbele binding van C=O. De Bindingslengten in de peptidegroep zijn aangegeven in Figuur 3. Vergelijk de C–N binding van de peptidegroep met die tussen N en Ca (het C-atoom waaraan de aminogroep en carboxylgroep zijn gehecht).

er zijn twee mogelijke conformaties van de vlakke peptidebinding: in de trans-peptidegroep bevinden de CA-atomen zich aan weerszijden van de peptidebinding (figuur 3a) en in de cis-peptidegroep bevinden de Ca-atomen zich aan dezelfde zijde van de peptidebinding (figuur 3b).
-
gezien de ruimtelijke ordening en de nabijheid van de atomen in het cis en trans-conformaties van de peptidebinding, welke conformatie zou volgens u de voorkeur krijgen?
-
de vervorming zou energetisch gunstiger zijn dan de dis-bevleesdheid, aangezien sterische hinder hierdoor tot een minimum wordt beperkt.
in het algemeen zijn peptidebindingen in de Trans-conformatie aanwezig. Nochtans, kunnen de cisvormen in peptide banden voorkomen die een Proline residu voorafgaan. In dergelijke gevallen is de cis-vorm stabieler dan gebruikelijk, omdat de Proline-zijketen minder hinderlijk is. Niettemin, komen de peptidebindingen van cis slechts in ongeveer 10% van gevallen van peptidebindingen voor die proline-residuen voorafgaan.
rekening houdend met de vlakke aard van de peptidegroep, kan een polypeptideketen een ruggengraat hebben die bestaat uit een reeks stijve vlakke peptidegroepen die met elkaar verbonden zijn door de Ca-atomen. Figuur 4 toont een deel van een polypeptide met twee vlakke peptide groepen in de Trans-Bouw. Merk op dat hoewel rotatie niet is toegestaan over de peptide bindingen, er een mogelijkheid is voor rotatie rond de Ca–N–en Ca-C-bindingen. De draaiingshoeken , genoemd torsiehoeken, over deze bindingen specificeren de bouw van een polypeptidebeenbeenderen. De torsiehoeken over de Ca–N-en Ca-C-bindingen worden ɸ (phi) en ψ genoemd. (psi), respectievelijk en zij worden gedefinieerd als 180° wanneer het polypeptide zich in de verlengde vlakke conformatie bevindt, zoals geïllustreerd in Figuur 4.
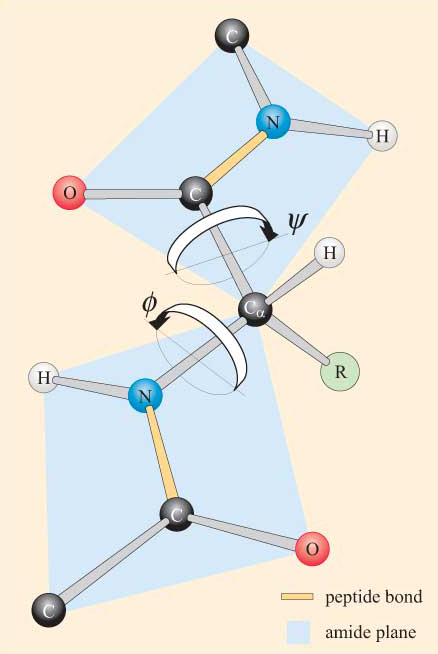
Het zal u niet verbazen dat sterische beperkingen van toepassing zijn op ɸ en ψ.
als gevolg van deze sterische beperkingen zijn alleen bepaalde waarden van ψ En ψ, en dus conformaties van het peptide, toegestaan, terwijl andere niet.
Het is mogelijk deze toegestane waarden voor een bepaald residu in de context van een polypeptide te berekenen. Deze berekening wordt uitgevoerd door eerst de afstanden te bepalen tussen alle niet-bindendeatomen in twee aangrenzende peptidegroepen (zoals die in Figuur 4) bij alle mogelijke waarden van ψ en ψ. Het wordt het gemakkelijkst gedaan voor een polypeptide die slechts één soort aminozuur bevatten. Een conformationele plot van ψ tegen ψ Voor een bepaald residu staat bekend als een plot van Ramachandran (naar zijn uitvinder, G. N. Ramachandran). Een dergelijke plot stelt ons in staat om die conformaties (dat wil zeggen voor een bepaalde waarde van ψ En ψ) Die sterisch gunstig of ongunstig zijn (zoals in Figuur 5) te identificeren aan de hand van de volgende criteria:
-
wanneer er geen conflict is tussen de van der Waals-stralen van niet-bindende atomen, is een conformatie ’toegestaan’. Deze conformaties liggen in de blauwe gebieden in Figuur 5.
-
conformaties die interatomische afstanden vereisen bij de grens van het toegestane gebied, worden gedefinieerd als “buitenste grens” – conformaties. Ze liggen in de groene gebieden in Figuur 5.
-
theoretische conformaties die vereisen dat twee niet-bindendeatomen dichter bij elkaar staan dan hun Van der Waals-stralen toestaan, zijn sterisch ‘verboden’. Deze liggen in de witte gebieden in Figuur 5.
merk op dat de waarden van ψ En ψ in Figuur 5 variëren van −180º tot +180º. Het draaien van de peptide groep door 360º zal het natuurlijk terug naar zijn beginpositie brengen, en −180º en +180º corresponderen met dezelfde positie. Zo is de groene strook in de linkerbenedenhoek van het perceel in Figuur 5 aaneengesloten met het veld in de linkerbovenhoek.
-
gebruik Figuur 5 om te bepalen of de volgende waarden van ɸ En ψ sterisch gunstig of ongunstig zijn: (a) ɸ = 90º en ψ = 90º; (b) ɸ = −90º en ψ = 90º.
-
(a) ongunstig; b) gunstig.
Ramachandran-plots kunnen worden geconstrueerd voor polymeren van elk van de 20 aminozuren. Het is belangrijk op te merken dat de Ramachandran percelen voor veel aminozuurresiduen over het algemeen zeer vergelijkbaar zijn, met slechts drie regio ‘ s met een gunstige of getolereerde conformaties (1-3 in de plot voor poly-L-alanine in Figuur 5). Verschillen doen zich echter voor. Bijvoorbeeld, waar de zijketen (R in Figuur 4) dicht bij Ca wordt vertakt, zoals in het geval van threonine, neemt het meer ruimte dicht bij de peptide backbone in en beperkt de benadering van atomen in de naburige peptide groepen. Dientengevolge, zijn toegelaten conformations (angles en ψ hoeken) beperkter voor polypeptides van vertakte aminozuren.
-
Proline verschilt ook sterk van andere aminozuren in termen van toegestane conformaties en voor polyproline worden alleen ɸ −waarden van −85º tot-35º getolereerd. Als je nadenkt over de structuur van proline, hoe kun je dan dit relatief smalle bereik van toegestane ɸ waarden verklaren?
-
de zijketen van proline is covalent gebonden aan de n van de aminogroep, dus in polyproline zal er minder rotatievrijheid zijn over de Ca–N binding dan met andere aminozuren. Bijgevolg zullen toegestane ɸ-waarden relatief beperkt zijn in vergelijking met andere aminozuren.
-
Figuur 6 toont de plot van Ramachandran voor glycineresiduen in een polypeptideketen. De regio ‘ s hebben een kleurcode zoals in Figuur 5. Wat kun je zeggen over de conformaties die glycine aanneemt? Denk aan de structuur van glycine. Waarom verschilt glycine van de andere residuen met betrekking tot zijn conformaties?
-
Glycine heeft een veel grotere conformationele vrijheid dan andere aminozuurresiduen, omdat het minder sterisch wordt gehinderd.

De plots van Ramachandran in de figuren 5 en 6 zijn gegenereerd voor respectievelijk L-alanine en L-glycine op basis van toegestane en buitenste grensafstanden voor interatomische contacten, bepaald op basis van bekende waarden voor Van der Waals-stralen van de atomen (Tabel 1).
Tabel 1 van der Waals afstanden voor interatomische contacten.
| contacttype | normaal toegestaan / Å | Outer limit / Å | H * * * H | 2.0 | 1.9 |
3.0 |
|---|
zij zijn daarom voorspellend in plaats van feitelijk conformationele plots. We kunnen natuurlijk röntgendiffractie gebruiken om experimenteel de ‘reële’ waarden van ɸ en ψ voor residuen in een polypeptide te bepalen. In Figuur 7 zijn de ɸ-En ψ-waarden voor alle residuen (met uitzondering van glycine en proline) in een aantal verschillende structuren bepaald door middel van röntgendiffractie met hoge resolutie en uitgezet op een Ramachandran-plot. We kunnen zien dat er een opvallende overeenkomst is tussen voorspelde en feitelijke conformaties. Merk echter op dat er een aantal residu ’s zijn waarvan de conformatie zich richt op de “verboden” gebieden. De meeste van deze residuen brengen de regio tussen de “toegestane” regio ’s 2 en 3 in kaart, rond ψ = 0.

-
kijk nog eens naar figuur 4 en stel je voor dat je de bovenste peptidegroep door 180° kunt draaien zodat ψ = 0. Welke groepen zijn waarschijnlijk in conflict in deze conformatie?
-
De N-H-groepen van aangrenzende peptide-groepen zullen met elkaar conflicteren en in de nabijheid worden gedwongen.
het conflict in verband met deze conformaties kan worden opgevangen door een kleine mate van verdraaiing van de peptidebinding. Aldus, in dergelijke conformations wordt de peptide groep uit zijn gebruikelijke vlakke Bouw gedraaid.
een beperkt aantal “verboden” conformaties van bepaalde residuen kan in een polypeptide worden getolereerd indien de aangenomen bevleesdheid als geheel energetisch gunstig is. Een polypeptide zal neigen zodanig te vouwen dat het de stabielste Bouw aanneemt. In deze Bouw minimaliseert het polypeptide zijn vrije energie. In de volgende secties zullen we kijken naar dit hogere niveau van eiwitstructuur.