- meghatározások és prevalencia
- Diagnózis
- Nccah megnyilvánulások nőknél
- megnyilvánulásai gyermekkorban
- serdülőkori és felnőttkori megnyilvánulások
- Nccah menedzsment az első megnyilvánulástól a felnőtt életig
- kezelés gyermekkorban
- kezelés serdülőkorban
- A gyermekkor óta kezelt betegek
- nők első megnyilvánulása serdülőkorban vagy hiperandrogén tünetek a kezelés abbahagyása után
- Nccah és Reproduction
- Subfertility
- termékenységi tervezés
- a CAH prenatális kezelése
- terhesség alatti kezelés
- egyéb speciális problémák
- stresszkezelés NCCAH-ban
- pszichobiológiai szempontok az NCCAH-ban
- következtetések
- szerzői hozzájárulások
- összeférhetetlenségi nyilatkozat
meghatározások és prevalencia
veleszületett mellékvese hiperplázia (CAH) magában foglalja a család autoszomális recesszív rendellenességek jellemzi enyhe vagy akutan károsodott kortizol szintézis hiánya miatt az egyik az öt mellékvese szteroidogén enzimek szükséges kortizol termelés (1, 2). Hagyományosan, CAH van osztva (a) klasszikus (CCAH), bemutató só-pazarlás vagy az egyszerű virilizáló formában, amely nyilvánvaló a születés és/vagy az újszülött időszakban, és (b) nem-klasszikus (NCCAH), ami egy kevésbé súlyos formája a betegség, amely nem rendelkezik a nemi kétértelműség, nem azonnal életveszélyes, és bemutatja később az élet, vagy tünetmentes marad, vagy téves diagnózis, mint egy másik betegség (3). A CAH különböző formáinak határait azonban nem szigorúan határozzák meg, ami növeli a rendellenességhez kapcsolódó kihívásokat. Ezért tanácsos a CAH-t a fenotípusok kontinuumának tekinteni, súlyostól enyhe vagy más tünetmentes (3).
a CAH leggyakoribb formáját a 21-hidroxiláz-hiány (21ohd) okozza, ami 17 hidroxiprogeszteron (17 OHP) 11 deoxikortizolra és progeszteron deoxikortikoszteronra történő átalakulását eredményezi. A szteroid konverzió blokádja az androgén prekurzorok fokozott termelését eredményezi, CRH-ACTH stimuláció alatt, ami biokémiai hiperandrogenizmushoz vezet, amelyet emelkedett 17 OHP szint jellemez. A betegség klasszikus formája világszerte 16 000 élveszületésből 1-ben fordul elő. Meg kell azonban említeni, hogy a betegség észlelt előfordulása az alkalmazott szűrési módszerhez kapcsolódik. Például arról számoltak be, hogy Svédországban a CAH klasszikus formájának előfordulása 1:11,500, ha esetfelmérési megközelítést alkalmaznak, ami azonban a hormonális szűrés során 1:9,800-ra csökken. Hasonlóképpen, Franciaországban és Svájcban az incidencia 1:15,472 és 1:23,000 és 1:10,749 és 1:11,661 között mozog, amikor a különböző szűrési módszereket alkalmazzák [4].
az NCCAH sokkal gyakoribb, 1000 kaukázusi közül körülbelül 1-nél fordul elő, és gyakrabban fordul elő bizonyos etnikai csoportokban, mint például az Askenazi zsidók (1:27), a spanyolok (1:53), a jugoszlávok (1:62) és az olaszok (1:300) (5, 6). Ennek ellenére hiányoznak a különböző becslési módszereken (esetfelmérés, hormonális szűrés, molekuláris tesztelés) alapuló nccah előfordulására vonatkozó adatok. Az NCCAH a leggyakoribb autoszomális recesszív endokrin rendellenesség, 1:25-1:10 vivőfrekvenciával. Az nccah-ban a 21-OHD miatt a maradék enzimatikus aktivitás becslések szerint körülbelül 10-70% (7-9).
Diagnózis
összehasonlítva a diagnózis a klasszikus formája a betegség, amely a szülés alatt, illetve újszülöttkori időszak, mert a nemi kettősség és/vagy só-pazarlás tünetek, vagy szűrési programok alkalmazott egyes országokban a legtöbb esetben a NCCAH nem könnyen kimutatható (4, 10). Továbbá, sok egyén tünetmentes marad gyermekkorban és serdülőkorban, normális reprodukciós funkcióval rendelkezik, és csak egy másik családtag diagnózisa és az azt követő tesztelés (11) miatt válik ismertté az NCCAH-ról. Az nccah-ban szenvedő nők többsége azonban orvosi segítséget kér, ha androgénfelesleg tüneteit tapasztalja, és amikor a klinikai gyanú vizsgálatot igényel, az emelkedett 17 OHP-szint valószínűleg nem mutat az NCCAH diagnózisára.
valójában az endokrin társadalom által javasolt klinikai iránymutatások 17 OHP kiindulási, nem stimulált értéket javasolnak az NCCAH szűrőeszközeként. Reggel 17 OHP szint >6 nmol/L a follikuláris fázisban a menstruáló nőknél további értékelést kell kérni, mivel kimutatták, hogy a 6 nmol/L feletti szintek az NCCAH egyének 90% – át (12, 13) rögzítik. A 17 OHP véletlenszerű mérése nem bizonyult hasznosnak, mivel ezek gyakran normális szintet eredményeznek az NCCAH-ban szenvedő betegeknél, ráadásul rendkívül magasak a luteális fázisban az egészséges nők felében (13). A 280 betegből származó adatainkban azonban hat beteg (2.1%) a kiindulási 17 OHP érték < 6 nmol/L (14) volt. Továbbá, bidé et al., a molekuláris technikákkal igazolt nccah-val rendelkező nők Nagy kohorszában a vizsgált alanyok 8% – ában (15) 6 nmol/L-nél alacsonyabb basal 17 OHP értékeket is találtak. Végül, a Speiser et al. által gyűjtött adatok alapján., Az nccah-val rendelkező egyének 9% – a 17 OHP értéket mutatott 2 ng/ml-nél alacsonyabb értéknél (ami 6nmol/L-nek felel meg). Más vizsgálatok szerint az nccah (13, 16, 17) diagnosztizálásához elegendő az 5, 1 és 9 nmol/L közötti kiindulási érték 17 OHP. A közelmúltban a bazális 17 OHP szintje 4.6 nmol/L-t javasoltak az ACTH-vizsgálat küszöbértékeként az nccah előrejelzésére gyermekkori korai adrenarche-ban szenvedő betegeknél (18).
ezeknek az eredményeknek és javaslatoknak az összege azt jelzi, hogy a Synacthen stimulációs teszten átesett betegek kiválasztását eseti alapon kell értékelni. A diagnózishoz 17 OHP utáni stimulációs szint szükséges 3 nmol/L felett (19). Az egy cyp21a2 mutációt hordozó heterozigóták ACTH stimuláció után enyhén emelkedett 17 OHP szintet mutatnak, bár átfedés van az érintetlen alanyokban (9). Azonban, mint Dacou-Voutetakis et al. rámutattak, hogy ha a bazális és posztstimulációs 17 OHP értékek összege meghaladja az 1,5 nmol/L értéket, akkor a heterozigozitás lehetősége rendkívül magas (20).
egy másik fontos szempont a 17 OHP értékeléshez használt technikák tekintetében. A 17 OHP-szintet különböző immunoassay-módszerekkel mérik, de amint azt a közelmúltban kimutatták, a legpontosabb és legmegbízhatóbb eredményeket a folyadékkromatográfia tömegspektrometriával (LC-MS/MS) történő kombinációjának alkalmazásával sikerült elérni. Valójában sok hamis pozitív 17 OHP mérést találtak, amikor az LC-MS / Ms méréseket összehasonlították a szokásos módszerekkel (21), azonban az utóbbi eljárásokat nem általánosan használják, bizonytalan diagnózis esetén pontosabb eredményeket nyújtanak. Megjegyzendő, hogy az LC-MS/MS számszerűsített 17 OHP-ra vonatkozó szűrési és diagnosztikai küszöbértékeket még meg kell határozni. Ezek a küszöbértékek az LC-MS/MS fokozott specificitása miatt valószínűleg alacsonyabbak, mint az immunoassay-kkel megállapítottak, amely módszer kevésbé hajlamos a keresztreaktivitásra és az interferenciákra (22, 23). Még nem tisztázott, hogy szükség van-e húgyúti szteroid profilra a végleges diagnózishoz (22). Borderline esetekben célszerű teljes adrenokortikális profilt szerezni az ACTH stimulációs teszt után, hogy megkülönböztessük a 21-hidroxiláz hiányt más enzimhibáktól, és szilárd diagnózist állítsunk fel.
specifikusan teljes szteroid profilt kell végezni kétértelmű esetekben a 21-hidroxiláz hiány igazolására és más enzimatikus kimutatások kizárására. 17 OHP, kortizol, 11-dezoxikortikoszteron,11-dezoxikortizol,17-OH-pregnenolon, dehidroepiandroszteron és androsztenedion felvétele a szérum szteroid profilba hasznos lenne a CAH egyéb okainak, például a 11β-hidroxiláz hiány, ritkábban a 3β-hidroxiszteroid-dehidrogenáz hiány vagy a P450 oxidoreduktázhiány (24) kizárására. Noha a genetikai vizsgálat jelenleg nem tekinthető az NCCAH elsődleges diagnosztikai eszközének, kötelező a diagnózis megerősítése és a genetikai tanácsadás (3).
ami a kortizolszintet illeti, általában legalább 496 nmol/L stimulációt követő érték várható. Megjegyzendő, szerint Stoupa et al., 60% – a 47 gyermekek NCCAH eredményeként 21 OHD volt alacsony kortizol értékek a stimuláció után, a megállapítás rámutatva, hogy szükség van a fokozott felügyelet a fejlesztés a mellékvese-elégtelenség során jelentős stresszor események (25).
Nccah megnyilvánulások nőknél
az nccah klinikai expresszióját a polimorfizmus magas szintje jellemzi, nemcsak a megjelenés korát, hanem a különböző jeleket és tüneteket is. Beszámoltak arról, hogy az NCCAH első klinikai bemutatása az esetek 11% – ában 10 éves kor előtt, 80% – ában pedig 10-40 éves kor között (12) történik. A CAH és az NCCAH genotípus-fenotípus összefüggése még nem tisztázott. Speiser et al. azt sugallják, hogy a 21-hidroxiláz hiány fenotípusos változékonyságának nagy része, de nem az összes, a CYP21A2 (26) allélos variációjából ered.
megnyilvánulásai gyermekkorban
az újszülött időszakban jellemzően az nccah nőstények tünetmentesek maradnak, és normális külső nemi szervük van. Az nccah legkorábbi esete egy 6 hónapos lány, aki fanszőrzetet fejlesztett ki (11). Az NCCAH-esetek klinikai eredményei és tünetei általában 5 éves kortól vagy még később kezdődnek, és az androgénszint emelkedésével kapcsolatosak, bár egyes esetekben enyhe kortizolhiány is előfordulhat (8). Az nccah-esetek 92% – ában az első tünet, amely a korai pubarche (PP), amely a lányoknál 8 év alatt, a fiúknál 9 év alatt fordul elő (12), becsült prevalenciája 10-11, 3% (15). Megjegyzendő, hogy a PP-vel említett gyermekeknél a különböző tanulmányok szerint az NCCAH előfordulása 5-30% (20, 27). Az nccah molekuláris megerősítésével rendelkező 280 beteg elemzése alapján azonban azt találtuk, hogy a PP előfordulása 94 8 évnél fiatalabb nőnél 88% volt. Másrészt az nccah-val rendelkező 45 férfi elemzése a PP-t csak az alanyok 29% – ában azonosította (14). Ezenkívül szem előtt kell tartanunk, hogy a PP-vel rendelkező alanyok körülbelül fele lehet A cyp21a2 mutáció heterozigóta hordozója.
egy másik szempont, amelyet tisztázni kell, a PP és a korai pubertás közötti kapcsolat. Feltételezzük, hogy bizonyos esetekben a mellékvese androgének ösztrogénekhez való perifériás aromatizálása aktiválhatja a hypothalamus-hipofízis-gonadális tengelyt, ami másodlagos korai pubertáshoz vezet (28). Ilyen esetben rendszeres nyomon követésre van szükség. Néhány gyermeknél kialakul a hirsutizmus, a pattanások és / vagy a gyors növekedés, valamint a magas struktúra és a csontkor előrehaladása (29). Ez utóbbi a gyors epiphysealis fúzió következtében csonka végső magasságot eredményezhet. A gyermekkori egyéb ritka klinikai eredmények közé tartozik a labiális adhézió, a perianális haj, a klitoromegália és a hang rekedtsége (30-32).
serdülőkori és felnőttkori megnyilvánulások
serdülőkorban és felnőttkorban az NCCAH nőknél a policisztás ovárium szindrómára (PCOS) emlékeztető hiperandrogén tüneteket mutat (33). A tünetek közé tartozik a hirsutizmus, akne, androgén alopecia, anovuláció, menstruációs zavar, meddőség. Egy multicentrikus vizsgálatban a serdülő és felnőtt nők körében a leggyakoribb tünetek a hirsutizmus (59%), az oligomenorrhea (54%) és az akne (33%) voltak. Az NCCAH-ban szenvedő 161 nő közül a tünetek a hirsutismus (78%), a menstruációs diszfunkció (54,7%) és a csökkent termékenység (12%) (34) voltak. A 161 betegből álló csoportunkban a beteg 63%-a policisztás petefészek-szerű fenotípusú (14). Ennek a megállapításnak fordítva van következménye, mivel az nccah előfordulása magas a hyperandrogenaemiában szenvedő nőknél. Természetesen a witcel et al tanulmánya. az nccah előfordulásának jelentése a hiperandrogén nőknél 1-33% között van. Egy másik vizsgálatban 950 nő, akiket az androgén felesleg értékelésére utaltak, az NCCAH-t 4, 2% – ában észlelték. Az NCCAH-betegek azonban fiatalabbak és hirsutabbak voltak a hiperandrogén betegek többi csoportjához képest. A tesztoszteron, a szabad tesztoszteron és a 17 OHP szintje is szignifikánsan magasabb volt, mint más hiperandrogén szindrómában szenvedő betegeknél. Ovarium ultrahang esetén 77% – uk policisztás petefészkeket, 41% – uk pedig fokozott ovariumméretet mutatott. Összességében 56, illetve 72,8% – os arányban teljesítették a NIH és Rotterdam szerinti PCOS-kritériumokat (35).
a betegség progresszív jellegét hangsúlyozza az a tény, hogy a hirsutizmus prevalenciája az életkorral növekszik, és megfigyelték, hogy a pubertás előtt ritka. A hirsutizmus a leggyakoribb klinikai megnyilvánulás, amelyet nccah-ban szenvedő betegeknél jelentettek, 71-96% (28, 36) között. Pubertás lányok NCCAH jellemzően jelen hirsutismus (37, 38). A spektrum másik végén az alopecia kérdése. Az nccah-ban szenvedő betegeknél férfi mintás kopaszodásról számoltak be. Az alopecia prevalenciája az életkorral is növekedett, a betegek 6% – ánál az életük második évtizedében 19% – ra az ötödikben, ami ismét jelzi a betegség progresszív jellegét (39). A pattanásokról az nccah-alanyok közel 33% – ánál (36) számoltak be. Figyelemre méltó, hogy az NCCAH-t okozó cyp21a2 gén mutációit a 123 felnőtt nő 4, 9% – ában észlelték súlyos pattanásokkal (40).
a menstruációs rendellenességek, beleértve az oligomenorrhea-t vagy a másodlagos amenorrhoeát, gyakran az nccah bemutató jele lehet a menarche utáni egyénekben. Ezenkívül az esetek 8-9% – ában elsődleges amenorrhoea is tapasztalható, ezért először orvoshoz kell fordulni. Ezenkívül az nccah egyének körében az oligomenorrhea gyakoribb, mint az elsődleges amenorrhoea. Például egy sor Moran et al., a 10-19 éves nccah nők körében az esetek 56% – ában oligomenorrhea volt jelen, szemben az elsődleges amenorrhoea (12) serdülőkorának csak 9% – ával. Végezetül, olyan sok nccah tünet hasonlít a PCOS-kre, hogy nem meglepő, hogy az NCCAH-t a PCOS nagy mimikerének nevezték el. Mindenesetre az nccah diagnózisát figyelembe kell venni minden olyan fiatal nő értékelése során, aki hiperandrogén tünetekre utal.
egy Arlt et al., az nccah-ban szenvedő betegeket szignifikánsan magasabb BMI és alacsonyabb végső magasság jellemezte a kiegyenlített kontrollokhoz képest (41). Ezek az adatok néhány, az inzulin érzéketlenségét jelző vizsgálattal együtt kedvezőtlen metabolikus profilra utalnak, amelyet kórélettanilag az emelkedett androgénkoncentrációk és/vagy a glükokortikoid terápia (42, 43) hatása magyarázhat. Csontritkulás is kimutatható ezekben az alanyokban, valószínűleg a kortikoszteroid terápia következtében (34).
Nccah menedzsment az első megnyilvánulástól a felnőtt életig
az NCCAH diagnózisának megállapítása után a későbbi kezelési ajánlásokkal kapcsolatos számos kérdés indokolja a megfontolást. Az első megválaszolandó kérdés az, hogy a glükokortikoid (GC) terápia javallt-e. Ennek a megközelítésnek az Általános érvelése az, hogy elegendő kortizol-koncentrációt biztosít a beteg számára, napi szükségleteit fedezi, következésképpen a CRH-ACTH tengely stimulációja kúpos lesz, ami csökkent mellékvese androgéntermeléshez vezet. E megközelítésre vonatkozó általános szabály az, hogy a GCs-t helyettesítő és nem farmakológiai dózisban adják, míg az életkor, a nem, a laboratóriumi adatok, a betegspecifikus ajánlások és a glükokortikoid-helyettesítő terápiára vonatkozó kezelési célok komolyan figyelembe vételével. Ugyanakkor tisztában kell lennünk azzal is, hogy a hosszan tartó GC szubsztitúciós terápia hypercortisolemia kialakulásához vezethet, az ebből eredő, jól dokumentált káros hatások az anyagcsere minden aspektusára, különösen a növekedésre, a zsíreloszlásra és az inzulinrezisztenciára, valamint a pszichológiai profilra. Ennek a megközelítésnek egy másik fő hátránya a megfelelő klinikai index vagy a megfelelő helyettesítő dózis biokémiai markerének hiánya,mint például a hypothyreosis TSH értékei.
az a tény, hogy nincs egyetértés abban, hogy melyik GC kell használni a hiánya a hosszú távú adatok tekintetében különböző módozatai GC adminisztráció tovább bonyolítják a részt vevő orvosok’ döntéseket kiválasztása az optimális választás minden beteg. A hidrokortizont általában gyermekeknél alkalmazzák, mivel leginkább a természetes hormonra (kortizolra) hasonlít, de serdülőknél és fiatal nőknél nem tekinthető megfelelő megközelítésnek, mivel többszörös napi adagolásra van szükség. Ezért a legtöbb felnőtt endokrinológus a dexametazont vagy a prednizolont részesíti előnyben megfelelő adagokban. Másrészt hangsúlyozni kell, hogy a különböző GC-k egyenértékűsége gyulladáscsökkentő hatásukon alapul, nem pedig az emberi anyagcsere különböző aspektusain. Mivel a kortizol az emberi genom közel 20% – át érinti, különböző szövetekben különböző GC-k különböző válaszai várhatók. Ezt az észlelést alátámasztja az a tény, hogy a dexametazon CAH-ban szenvedő betegeknél történő alkalmazása az inzulinrezisztencia indexeinek romlását mutatta más GCs-hez képest (44). Ezenkívül a 2,5-7,5 mg prednizolon adagolása, amely a normálisnak tekintett dózis, régóta negatív hatást gyakorol a csont anyagcserére (45). Ezért a hidrokortizont a kezelés legjobb formájának kell tekinteni GC kiegészítő terápia esetén. Az újonnan szintetizált hidrokortizon formula napi egy tablettával való megjelenése, valamint a CAH-ban szenvedő betegek kezdeti pozitív eredményei sok ígéretet mutatnak a jövőre nézve (46).
kezelés gyermekkorban
NCCAH a gyermekkorban PP jelekkel diagnosztizált betegeket hidrokortizonnal lehet kezelni a mellékvese hormonok elnyomása és a csontkor gyors előrehaladásának megakadályozása érdekében, amely befolyásolhatja a végső magasságot. Azoknál a betegeknél, akiknél a hidrokortizon-kezelést 1 évvel a pubertás kezdete előtt kezdték meg, és akiknél a csont életkora 9 év alatt volt, a végső magasság a genetikai potenciálon belül maradt (47). A rendelkezésre álló vizsgálatok azt mutatják, hogy a felnőtt testmagasság megközelítette a várt célmagasságot azoknál a betegeknél, akiket szoros megfigyelés alatt tartottak, és akik szigorúan betartották a gyógyszerterveket (48-50). Egy nemrégiben készült, öt esetből álló kis vizsgálat (6,1–9,2 éves betegek) kimutatta, hogy a dexametazon ultralow adagja ígéretes lehetőség az endogén stressz és annak hatásainak csökkentésére. Annak tisztázása, hogy ez reális megközelítést jelent-e, és hogy a beadott dózist mennyiben kell meghatározni (51). A tanulmány szerint Nebesio et al. a hidrokortizon, prednizolon és dexametazon hormonális hatásait és farmakokinetikáját hasonlította össze 9 CCAH-ban szenvedő prepubertális betegnél. Kimutatták, hogy a dexametazon hatásosabb, mivel a többi forma lényegesen alacsonyabb mellékvese hormonszint elérését teszi lehetővé, ami arra utal, hogy a dexametazon hatásosabb a mellékvese androgén termelésének elnyomására (46). Természetesen további vizsgálatokra van szükség e megállapítás ellenőrzéséhez vagy elutasításához. Ezenkívül egyes esetekben az emelkedett androgén koncentráció a GnRH tengely másodlagos stimulálásához vezethet, ami korai pubertáshoz vezet. Ebben az esetben a GnRH analógdal párhuzamos kurzust lehet előírni, ha a csontkor lényegesen magasabb, mint a kronológiai kor, és/vagy a tervezett végső magasság aránytalan a célmagassághoz képest.
egy másik fontos szempont az egyidejű GH-hiány eseteit illetően abban az esetben, ha a GnRH-analógokkal és GH-receptre kapható gyógyszerekkel történő kezelés a célmagasság elérését eredményezte (52). A jelenlegi iránymutatások szerint a fent leírt terápia csak olyan esetekben ajánlott, amikor az SD várható magassága -2, 25 < a célmagasság vagy még alacsonyabb (53). A hidrokortizon-kezelés megkezdésének alternatív indikációja az ACTH-stimuláció utáni nem megfelelő kortizol válasz (36).
gyermekeknél az előnyben részesített GC-kezelés általában 10-15 mg/m2 hidrokortizon, három adagra osztva. Gyakran az alacsonyabb dózisok is hatékonynak bizonyultak, kezdve 6 mg/m2/nap (34). A túladagolást kerülni kell, figyelembe véve, hogy ez rossz növekedést, valamint Cushingoid tulajdonságokat eredményezhet. A rendszeres növekedési mintázat, az időrendi korral összeegyeztethető csontkor, valamint a központi elhízás hiánya klinikai mutatóként is szolgálhat a megfelelő kezeléshez. Kapcsolatos biokémiai/ hormonális profil androsztendion, valamint a tesztoszteron szintje a közepén a felső tartományban a csont-kor tartják jobb markerek a megfelelő GC pótlás a gyermekek NCCAH. Az nccah-ban szenvedő betegek 10% – ánál azonban elnyomott tesztoszteronszintet találtak, míg a betegek további 28% – ánál nőtt a tesztoszteron koncentrációja, ez a jelenség valószínűleg a hidrokortizon variabilitásának tulajdonítható (54, 55). A DHEAS-értékek nagyon kis dózisokkal csökkennek, míg a 17 OHP és a progeszteron szint elnyomása nagyon magas GC-dózisokat igényel. Meg kell jegyezni, hogy 17 OHP érték döntő fontosságú a diagnózis szempontjából, de nem hasznos a nyomon követés során. Megjegyzendő, hogy a hormonális értékeléshez szükséges vérmintát a terápia abbahagyása nélkül kell elvégezni. A betegség zavarossága és sokrétű jellege miatt azonban nincsenek konkrét iránymutatások a kezelési rend változásainak időzítésére vagy a glükokortikoid terápia abbahagyására gyermekeknél.
kezelés serdülőkorban
A gyermekkor óta kezelt betegek
amíg az nccah lányok normális menstruációs mintája meg nem állapodik, a gyermekkorban kezdődő GCs folytatása erősen ajánlott (56). A serdülő betegek azonban gyakran nem mutatnak kellő mértékű megfelelést a gyógyszerek krónikus adagolásának, és gyakran kihagyják az adagokat. Ezenkívül a pubertás alatt a hidrokortizon felezési ideje 50%-kal csökken a megnövekedett IGF-1 szintek következtében, ami csökkenti a 11ßohsd aktivitást, valamint a glomeruláris filtrációs sebesség erősítéséből eredő megnövekedett kortizol clearance miatt (57). A fiatal nősténynél a menarche után 2 évvel, és ha normál ovulációs ciklusokat regisztráltak, erősen ajánlott a betegközpontú megközelítés a megjelenő hiperandrogén tünetek felé.
ha súlyos hiperandrogén eredmények vannak, például hirsutizmus, akne és/vagy oligomenorrhea, a kezelés folytatását mérlegelni kell. Fontos megjegyezni egy fiatal serdülő törékeny érzékenységét, amelyet súlyosan károsítanak a hiperandrogenizmus “visszataszító” jelei. A hiperandrogén jelek, a menstruációs rendellenességek és a rossz életminőség kombinációja jól dokumentált, és különösen magas a fiatalabb nőknél. Egy másik szempont, amelyet figyelembe kell venni, a mellékvese rezervátum reakciója az ACTH stimuláció után. Tekintettel arra, hogy a pubertás kortizol csúcsértéke és a stimuláció után a felnőtt nők kortizolszintje 496 nmol/L alatt van, stressz esetén szteroid kezelést kell alkalmazni (58). Másrészről, ha a fenti tünetek egyike sem jelentkezik a fiatal betegnél, a GC-kezelés megszakítható, majd rendszeres nyomon követés javasolt.
nők első megnyilvánulása serdülőkorban vagy hiperandrogén tünetek a kezelés abbahagyása után
hiperandrogén tünetek jelenlétében erősen ajánlott betegorientált megközelítés, a beteg fő panaszaira összpontosítva. Ez sokkal jobb, mint egy általános univerzális működési mód, mivel ez utóbbi megközelítéssel a fiatal beteg csalódott lesz, ami a kezelés abbahagyásához és ennek eredményeként a felnőttkorban káros egészségügyi eredményekhez vezet. Súlyos hirsutizmus esetén az orális fogamzásgátló tablettákkal (OCPs) végzett 6-12 hónapos kezelés ésszerű eljárást jelent, mivel az OCPs jótékony hatással van az SHBG májtermelésre és az androgén felszabadulás csökkenésére a petefészekből. Legalább 2-3 hónapos OCPs-kezelés megkezdése után klinikai javulás várható, míg minimális androgén tulajdonságokkal rendelkező progesztagén egyidejű alkalmazása erősen ajánlott. Ha az OCP-k az első vonalbeli megközelítés során kudarcot vallanak, antiandrogének (spironolakton, flutamid, bikalutamid, ciproteron-acetát és finaszterid) adhatók a kezeléshez. Tapasztalataink szerint a bikalutamid alkalmazása jelentős javulást ért el súlyos akne esetén, de hasonló eredményeket nem kaptunk súlyos hirsutizmus esetén. Kozmetikai megközelítések, mint a lézeres alkalmazás és szőrtelenítők is javasolható a nők panaszkodnak a túlzott vagy nem kívánt haj növekedését vagy fejbőr hajhullás (androgén alopecia) (32, 34, 57).
azoknál a betegeknél, akiknél a stimuláció után nem megfelelő a kortizolszekréció, vagy ha az OCPs és az antiandrogének nem tolerálhatók, erősen ajánlott a GCs-kezelés (57). Adatok New et al. jelezze, hogy a glükokortikoid-kezelés megkezdését követő 3 hónapon belül a szabálytalan menzesz és akne megfordul,míg a hirsutizmus közel 30 hónapot igényel (59). A gyermekkorral ellentétben serdülőkorban gyakran alkalmaznak hosszabb hatású szteroidokat, 5 mg prednizolon vagy 0,25 mg dexametazon (53) ajánlott. A valós világban azonban a klinikai adatok számos különböző kezelési módot mutattak az NCCAH menedzsmentben (41).
a kezelés elsődleges célja a páciensnek a hiperandrogén tünetek enyhítése. Így a kizárólag laboratóriumi eredmények alapján diagnosztizált nccah-val rendelkező egyének körében a klinikai jellemzőknek vezetniük kell a kezelési ajánlásokat, mivel a hormonális és molekuláris eredmények nem feltétlenül jelzik a klinikai súlyosságot. Az endokrin társadalomra vonatkozó iránymutatások szerint az Nccah-ban szenvedő betegeknek lehetőséget kell biztosítani a GC-kezelés abbahagyására, amikor a tünetek megszűnnek (60). Természetesen ezeket a betegeket nem szabad elveszíteni a nyomon követés során, míg a kezelést a tünetek megismétlődése esetén újra kell kezdeni. Továbbá a kezelés abbahagyása esetén a betegeket tájékoztatni kell a meddőség lehetőségéről, és ösztönözni kell őket arra, hogy ha teherbe kívánnak esni, forduljanak orvoshoz (19). Megjegyzendő, hogy a páciensnek a gyermekgyógyászról a felnőtt endokrinológusra történő megfelelő átvitelét optimálisan kell elvégezni 1 éves szinkronizált monitorozás után (53, 61).
Nccah és Reproduction
Subfertility
a Szubfertilitást gyakran jelentik a CAH klasszikus formáiban, ami elsősorban a menstruációs rendellenességeknek, a krónikus anovulációnak és az anatómiai deformitásoknak köszönhető. A születési arányt 17% – ra becsülték, szemben a kontrollpopuláció 65% – ával (62). Eközben az nccah-ban szenvedő nők termékenységére vonatkozó adatokat a közelmúltban részletesen megvizsgálták, és az nccah-ban szenvedő nők becsült meddőségi incidenciája 11%, azaz viszonylag enyhébb, mint a CAH-ban, és sok esetben könnyen megoldható. Bidé et al. az nccah-ban szenvedő 190 nőnél értékelték a termékenységet, akik közül 95 teherbe akart esni. Ebben a populációban 85 nőből 187 terhességről számoltak be, ami 82 személynél 141 születést eredményezett. Ki kell emelni, hogy a terhességek 99-E (52, 9%) az NCCAH diagnózisa előtt történt, közülük három ovuláció indukciós protokollok alkalmazásával történt, a többi spontán, míg az NCCAH diagnózis után 88 terhesség történt. Túlnyomó többségükben a terhesség a hidrokortizon terápia megkezdése után alakult ki, míg 11 nőnél spontán történt (63, 64). Egy másik jelentésben, amelyben 22 megfigyelt nccah-nő kívánt terhességet, 12 terhesség következett be prednizonnal (65).
a bidet et al., a vetélések aránya 6,5, illetve 26,3% volt a GCs-vel kezelt és kezeletlen betegeknél (64). A terhesség kimenetele glükokortikoid kezelés nélkül is sikeres lehet azokban az esetekben, amikor az NCCAH-t még nem diagnosztizálták, amint azt a Cuhaci et al. Ennek ellenére ugyanaz a pár két vetélést tapasztalt, és az első terhesség után (66) szubfertilitásról számolt be. Ami a koraszülött terhességek lehetőségét illeti, a bidet et al., nem különbözik jelentősen a lakosság többi részétől (15).
termékenységi tervezés
azoknál a nőknél, akiknél tünetekkel járó hiperandrogenizmus vagy jelentett meddőség áll fenn, de akik teherbe kívánnak esni, a GC-kezelés erősen ajánlott. Ebben az időszakban az alkalmazott glükokortikoidok prednizon vagy hidrokortizon. A legfontosabb intézkedés, mivel a következő lépéseket irányítja, a leendő szülők genetikai tesztelése. Az nccah-val rendelkező nőknek tisztában kell lenniük a betegség klasszikus formájával rendelkező csecsemő születésének kockázatával. Az nccah-ban szenvedő nők terhességének kimenetelét, pontosabban a CCAH-val született csecsemők incidenciáját a közelmúltban végzett vizsgálatok 1, 5-2, 5% – ra becsülik, az NCCAH pedig körülbelül 15% (36, 64). Természetesen ez a gyakoriság a referenciapopulációtól függ, magasabb előfordulási gyakoriság a magas házasságkötési arányú populációkban (36). A CAH-val rendelkező gyermek szülésének várható esélye ismeretlen apai genotípus esetén 1: 120, nulla, ha az apának nincs releváns mutációja, 1: 4, Ha heterozigóta mutációja van (19). Ennek eredményeképpen ezeknek a nőknek a partnereinek genetikai vizsgálata elengedhetetlen a CAH (64, 67) klasszikus formájával rendelkező gyermek születésének kockázatának felméréséhez.
Abban az esetben, ha a leendő anya hordozza a két NC mutációk (1. Táblázat), nem feltétlen a genetikai vizsgálatok a jövőben apja, mivel, ha heterozigóta a CYP12A2 mutációk, a lény kialakulásának kockázata a nem-klasszikus formája a betegség a jövőben. Meg kell azonban jegyezni, hogy a hordozó státusz 2-15% a különböző populációkban, ezeknek az egyéneknek a fele súlyos mutációt hordoz. Ezenkívül a hiányos genotípus-fenotípus korreláció miatt nehéz végleges ajánlásokat tenni olyan helyzetekre vonatkozóan, amikor a genetikai vizsgálat nem szükséges, különösen az enyhébb mutációk esetében . Például a p30l mutáció, amely leggyakrabban az NCCAH-val társul, a klasszikus CAH-ban szenvedő betegeknél is előfordul . Egy nagy multicentrikus európai tanulmány a közelmúltban kimutatta, hogy ez a helyzet a betegek 78% – ánál . Ebben a vizsgálatban az enyhe v218l mutáció az esetek 30% – ában az Nccah helyett a klasszikushoz kapcsolódott. Ezért minden nccah-ban szenvedő betegnek genetikai tanácsadást és a reproduktív partnerek molekuláris értékelését kell felajánlani.
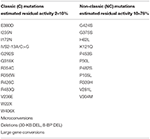
1. táblázat. A cyp21a2 mutációi minimális (C) vagy mérsékelt (NC) maradék enzimaktivitással (14, 68, 69).
ezzel szemben abban az esetben, ha a leendő anya egy összetett heterozigóta, amelynek egy súlyos (C) és egy NC mutációja van, akkor a jövőbeli apa genetikai vizsgálata kötelező. Szem előtt kell tartanunk, hogy az immunoassay-kkel vagy a pontosabb LC-MS/MS-eljárásokkal végzett vizsgálatokból származó adatok következetesen kiemelik azt a képtelenséget, hogy megbízhatóan kizárják a heterozigozitást bazális vagy ACTH stimulált 17 OHP értékekkel, a normál tartomány jelentős átfedése miatt. Az egyik egy Szinaktén teszt alkalmazását javasolhatja, és ha nem kompatibilis a heterozigozitással (a bazális és csúcs stimulált 17 OHP értékek összege < 1, 5 nmol/L), akkor a DNS-vizsgálat elkerülhető. Ennek a tesztnek az értelmezésében azonban nagyon óvatosnak kell lennünk,mivel más vizsgálatokban nem érvényesítették. Ezért az ACTH stimulált 21-deoxycortisol alkalmazása akár külön-külön, akár szteroid Arány vagy szteroid profil részeként megkönnyítheti a heterozigóták biokémiai azonosítását a jövőben, különösen mivel az LC-MS/MS szélesebb körben elérhetővé válik . Ha a leendő apa NC mutációt hordoz, akkor semmi másra nincs szükség. Ezzel szemben, ha egy klasszikus mutáció hordozója, felmerül a kérdés a magzat prenatális kezelésével kapcsolatban. Az összes lehetséges genotípus-kombináció és a javasolt eljárások a 2. táblázatban találhatók.

2. táblázat. A javasolt eljárások tervezése a terhesség alatt a leendő szülők genotípusa alapján.
a CAH prenatális kezelése
a CAH prenatális kezelésének célja a magzat genitális virilizációjának megelőzése, de a szülők által érzett stressz enyhítése is, akik valószínűleg kétértelmű nemi szervekkel rendelkeznek (70). A dexametazont azért alkalmazzák, mert képes átjutni a placentán, és mivel nem deaktiválódik a placenta 11β Oh szteroid dehidrogenázából, és csak minimálisan kötődik az anya véréhez kortizolkötő globulinhoz (CBG). A dexametazon a magzati ACTH szekréció elnyomásával csökkenti az emelkedett magzati androgén termelést. A kezelést abba kell hagyni abban az esetben, ha a kariotípus vagy a DNS-elemzés férfi vagy nem érintett nőt tár fel (71). Mindazonáltal, bár a magzati genitális virilizáció már a fogantatás után 6-7 héttel kezdődött, a genetikai diagnózishoz szükséges korionos villous biopsziák nem szerezhetők be a 10.-12. hét előtt. Ez az időintervallum azt sugallja, hogy a virilizáló CAH-t veszélyeztető összes terhességet kezelni kell, annak ellenére, hogy a 8 magzatból csak 1 érintett és nő (19, 34, 70).
továbbra is ellentmondásos, hogy a magzat dexametazonnak való kitettsége megelőző intézkedésekhez orvosilag és etikailag elfogadható-e (70). Állatkísérletek igazolták, hogy az első trimeszterben dexametazon csökken súllyal befolyásolja a vese-hasnyálmirigy béta-sejt működés, valamint az agy fejlődését, valamint veheti szorongás, magas vérnyomás, valamint hyperglykaemia a felnőttkorban (70). Továbbá, 10-20% – a terhes nők a dexametazon kezelés, a terhesség alatt panaszkodnak a súlygyarapodás, fokozott étvágy, hangulatingadozás, álmatlanság, edemas, magas vérnyomás, hyperglykaemia, valamint a barázdák. Azonban új et al. a dexametazonnal kezelt magzatokban az átlagosnál alacsonyabb Prader-pontszámot találtak, de a hosszú távú eredmények között nincs különbség (72). Továbbá, a felülvizsgálat Merce Fernandez-Balsells et al. kimutatták, hogy a dexametazon a magzat virilizációjának csökkenésével jár, jelentős anyai vagy magzati káros hatások nélkül (63). A fent említett felülvizsgálat szerzői, valamint az endokrin társadalom egymást követő iránymutatásai azonban rámutatnak arra, hogy a szakirodalom egész testében található kis mintaméretek miatt a téma továbbra is bizonytalan, és egyértelműen további vizsgálatra van szükség.
A döntést arról, hogy a kezelés megkezdése csak akkor kell végezni, nagy központok egy tapasztalt csapat protokollok által jóváhagyott Intézményi Felülvizsgálat Táblák alapján a családi értékeket, beállítások, valamint az írásbeli tájékoztatáson alapuló mint előfeltétel (19, 63). Több szakemberek a mező jelzi, hogy a magasabb értéket kell helyezni megakadályozza a felesleges prenatális expozíció az anya, mind a magzat, hogy a dexametazon ahelyett, impozáns, az érzelmi veszteség kétértelmű nemi szervét a szülők, mind a betegek. A legfontosabb, hogy tisztázni kell a szülőknek, hogy a dexametazon alkalmazása nem módosítja a beteg állapotát, hanem a műtét szükségességének csökkentésére irányul, nem pedig az élet vagy az intellektuális kapacitás megőrzésére. Azt is tudniuk kell, hogy a kezelés ellenére nagy a valószínűsége annak, hogy gyermekük a betegség klasszikus formájától szenved. A legfontosabb, hogy eközben egy fiatal szervezet nagyon erős GC-nek való kitettsége a magzati programozás és növekedés különösen érzékeny időszakában, amely a XY kariotípusú magzat esetében haszontalannak bizonyulhat, jelenleg nem támasztják alá robusztus és megkérdőjelezhetetlen adatok.
ideális esetben korai diagnózisra van szükség, ezt nem invazív technikával, a magzat genitális virilizációjának kezdete előtt kell elvégezni. Ebben az irányban dolgozva az új vizsgálatok arra utalnak, hogy az anyai plazmából nyert sejtmentes magzati DNS-t ígéretes módszerként alkalmazzák, amely lehetővé teszi a magzati nem meghatározását és a CAH korai terhességi korban történő diagnosztizálását (<9 hét). Ha ezt az eljárást széles körben alkalmazzák a klinikai gyakorlatban, elkerülhető a felesleges prenatális dexametazon kezelés (73).
terhesség alatti kezelés
terhesség alatt az NCCAH-ban szenvedő nőket lehetőleg hidrokortizonnal kell kezelni, nem csak azért, mert a terhességet általában magas stresszszint kíséri, hanem megelőző intézkedésként is a fent említett magas vetélés előfordulása ellen. A glükokortikoid ezen formáját előnyben részesítik a placenta 11β Oh szteroid dehidrogenáz II enzim általi metabolizációja miatt, amely megakadályozza, hogy a glükokortikoid bármilyen hatással legyen a magzatra (71). A fogantatás idején a hidrokortizon adagját napi 20-25 mg-ra kell növelni, az adagot pedig 6-8 hetente kell módosítani annak érdekében, hogy a tesztoszteronszintet a terhesség trimeszterének felső normál szintjén tartsák (74). Példaként az nccah-ban szenvedő beteg tesztoszteron-és D4-értékeit mutatjuk be a fogantatástól a két egészséges ikrek szülés utáni 3 évig (1. ábra).
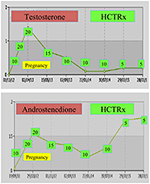
1.ábra. A tesztoszteron és az andosztenedion szintje és az ezt követő hidrokortizon adagolása (HCTRx) a fogantatástól az nccah-ban szenvedő betegnek történő beadást követő 3 évig.
egyéb speciális problémák
stresszkezelés NCCAH-ban
súlyos életveszélyes stressz, műtét vagy súlyos betegség esetén a glükokortikoiddal kezelt nccah-ban szenvedő betegeknek nagyobb vagy gyakoribb glükokortikoid dózisokra lehet szükségük, mivel mellékvese funkciójuk iatrogenikusan elnyomódik. Ezért elengedhetetlen a kisgyermekek szüleinek oktatása, valamint a betegek oktatása a felnőttkori gondozásra való áttérésről, a stressz adagolásáról. Azoknál az NCCAH-s betegeknél, akiket nem kezelnek GCs-vel vagy abbahagyás esetén, meg kell vizsgálni az ACTH-re adott kortizolválaszt. Az NCCAH betegek csaknem egyharmada nem megfelelően reagál a stimulációra (15, 75). Azok számára, akik normálisan reagálnak az ACTH stimulációra, nem ajánlott stresszadagokkal kezelni (19). A mineralokortikoid terápia egyik esetben sem szükséges, mivel ezek a betegek normális aldoszteron szekrécióval rendelkeznek, és nem alakulnak ki sópazarlás (58). Megjegyzendő, hogy hyperthyreosis vagy a levotiroxin kezelés növekedése esetén a kortizol clearance megemelkedik, mellékvese válság léphet fel (58, 76).
pszichobiológiai szempontok az NCCAH-ban
Meyer-Bahlburg et al. számos tanulmányban beszámoltak az nccah-ban szenvedő betegek pszichológiai profiljának károsodásáról a 21OH-hiány miatt. Pontosabban, az érintett nők magasabb masculinization / defeminization pontszámot kaptak a nemekkel kapcsolatos viselkedés számos intézkedésénél, összehasonlítva a normál kontroll nőkkel, bár jelentősen kevésbé, mint a klasszikus CAH-ban szenvedő nőknél. Jelentősen megnövelték a biszexuális vagy homoszexuális orientációt is. Retrospektív klinikai-kvalitatív interjúk ezek a nők kiderült, a kórtörténetben a kellemetlen érzés és a társadalmi stressz kapcsolatos a kezelés előtti tapasztalatok androgén-függő tünetek, mint például a pattanások, hirsutismus, és koncepció nehézségek (59, 77, 78). Hasonlóképpen, Arlt et al. azt mutatta, hogy a szubjektív egészségi állapot jelentősen csökkent mind a nyolc területen egy rövid távú egészségügyi felmérés, a legjelentősebb különbségek, mint a kor-és szex-kiegyenlített ellenőrzések, kapcsolatos területek általános egészségügyi, vitalitás, szerep korlátozások miatt érzelmi problémák. A kórházi szorongás és depresszió skála (HADS) kérdőív szintén fokozott szorongási pontszámokat tárt fel (41). Figyelembe véve ezeket a megállapításokat, az NCCAH-ban szenvedő nőknél figyelembe kell venni a terápia irányításának pszichológiai paramétereit, és a betegorientált megközelítés összefüggésében pszichológiai diagnózist és támogatást kell kínálni.
következtetések
az nccah a CAH gyakoribb és enyhébb formája a 20-50% – os enzimaktivitás megtartása miatt. A legtöbb beteg életük bármely szakaszában orvoshoz fordulhat az androgén felesleggel kapcsolatos tünetek miatt. Ezek közé tartozik a korai pubarche, a gyors növekedés, a hirsutizmus, a pattanások, a menstruációs rendellenességek vagy a meddőség, valamint számos más, kevésbé kiemelkedő megnyilvánulás. A kora reggeli alapértékek 17 OHP, mint egy jó kezdeti szűrővizsgálat és további értékelés ACTH stimulációval, valamint borderline eredmények esetén genetikai vizsgálat ajánlott. Általános szabályként az androsztén-Diont és nem a 17 OHP-szintet kell alkalmazni a nyomon követés során.
a kezelési határozatnak a tények értékelésén kell alapulnia, és egyénre szabott megközelítést kell követnie. A kezelés nem mindig javallt, kivéve, ha a beteg tüneti jellegű, például a korai kezdetű és gyors progresszióját szemérem-és testszőrzet, gyors növekedés,és / vagy csontváz haladás, vagy a nők oligomenorrhea, akne, hirsutismus, meddőség, vagy ezek és mások kombinációja a fent említett tünetek. Genetikai tanácsadás erősen ajánlott nccah nők, akik szeretnék elképzelni, valamint genotipizálás az apa. A beteg általános kezelése magában foglalja a glükokortikoid terápia valószínű szövődményeinek kezelését vagy a betegség anyagcserével kapcsolatos megnyilvánulásait is. Végül, tekintettel arra, hogy e betegek megfelelő kezelésével kapcsolatos számos terápiás kérdés még nem tisztázódott, nagyon fontos, hogy a kezelőorvos naprakész legyen az ezen a területen bekövetkezett összes fejleményről, és integrálja az új adatokat klinikai gyakorlatába. Természetesen további klinikai vizsgálatok ezen a területen elengedhetetlenek.
összefoglalva, a NCCAH nő, az ideális a megközelítés egy személyre szabott egyik, amely a zökkenőmentes átmenetet a menedzsment egyszer, hogy az említett, a gyermek a felnőtt endokrinológus, valamint a tünet-orientált kezelés, ami elkíséri egész életében. Tekintettel a hiperandrogén rendszert befolyásoló több tényezőre, tanácsos ösztönözni a betegeket, hogy egészségesebb életmódot fogadjanak el étkezési szokásaik javításával, a testmozgás növelésével, valamint a súlycsökkentés céljával. Ezenkívül bizonyos esetekben a pszichológiai támogatás gyakran előnyös. Ezen túlmenően a betegség kezelésében részt vevő szakemberek, például bőrgyógyászok, nőgyógyászok, pszichológusok, mélyreható megértést igényelnek az nccah gyanús vagy már diagnosztizált eseteinek kezeléséről. Végezetül tartsuk szem előtt Vera Cooper Rubin amerikai csillagász éleslátó idézetét: “a tudomány a legjobban halad, amikor a megfigyelések arra kényszerítenek bennünket, hogy megváltoztassuk előítéleteinket.”
szerzői hozzájárulások
minden felsorolt szerző jelentős, közvetlen és intellektuális hozzájárulást nyújtott a munkához, és jóváhagyta közzétételre.
összeférhetetlenségi nyilatkozat
a szerzők kijelentik, hogy a kutatást olyan kereskedelmi vagy pénzügyi kapcsolatok hiányában végezték, amelyek potenciális összeférhetetlenségnek tekinthetők.
1. Speiser PW, fehér PC. Veleszületett mellékvese hiperplázia. N Engl J Med. (2003) 349:776–88. doi: 10.1056/NEJMra021561
PubMed Abstract / CrossRef teljes szöveg / Google Scholar
2. Turcu AF, Auchus RJ. Adrenal steroidogenesis and congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. (2015) 44:275–96. doi: 10.1016/j.ecl.2015.02.002
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Speiser PW. Nonclassic adrenal hyperplasia. Rev Endocr Metab Disord. (2009) 10:77–82. doi: 10.1007/s11154-008-9097-x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Gidlöf S, Falhammar H, Thilén A, von Döbeln U, Ritzén M, Wedell A, et al. Száz éves veleszületett mellékvese hiperplázia Svédországban: retrospektív, populációs alapú kohorsz vizsgálat. Lancet Diabetes Endocrinol. (2013) 1:35–42. doi: 10.1016/S2213-8587(13)70007-X
PubMed Abstract | CrossRef teljes szöveg | Google Scholar
5. Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan a, Új MI. A nem klasszikus szteroid 21-hidroxiláz hiányának nagy gyakorisága. J Hum Genet Vagyok. (1985) 37:650–67.
PubMed Abstract | Google Scholar
6. Wilson RC, Nimkarn S, Dumic M, Obeid J, Azar MR, Azar M, et al. Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Mol Genet Metab. (2007) 90:414–21. doi: 10.1016/j.ymgme.2006.12.005
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Miller WL. Clinical review 54: genetics, diagnosis, and management of 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1994) 78:241–6. doi: 10.1210/jcem.78.2.8106606
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Witchel SF, Azziz R. Nonclassic congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2010) 2010:625105. doi: 10.1186/1687-9856-2010-625105
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Wilson RC, Mercado AB, Cheng KC, Új MI. Szteroid 21-hidroxiláz hiány: a genotípus nem tudja megjósolni a fenotípust. J Clin Endocrinol Metab. (1995) 80:2322–9. doi: 10.1210 / jc.80.8.2322
CrossRef teljes szöveg | Google Scholar
10. Fehér PC. Újszülöttkori szűrés veleszületett mellékvese hiperplázia esetén. Nat Rev Endocrinol. (2009) 5:490–8. doi: 10.1038 / nrendo.2009.148
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Kohn B, Levine LS, Pollack MS, Pang S, Lorenzen F, Levy D, et al. Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. (1982) 55:817–27. doi: 10.1210/jcem-55-5-817
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Moran C, Azziz R, Carmina E, Dewailly D, Fruzsetti F, Ibañez L, et al. A 21-hidroxiláz-hiányos nemklasszikus mellékvese hiperplázia progresszív rendellenesség: multicentrikus vizsgálat. Am J Szülész Nőgyógyász. (2000) 183:1468–74. doi: 10.1067 / mob . 2000.108020
PubMed Abstract | CrossRef teljes szöveg | Google Scholar
13. Azziz R, Hincapie LA, Knochenhauer ES, Dewailly D, Fox L, Boots LR. 21-hidroxiláz-hiányos nemklasszikus mellékvese hiperplázia szűrése hiperandrogén nők körében: prospektív vizsgálat. Fertil Steril. (1999) 72:915–25. doi: 10.1016/S0015-0282(99)00383-0
PubMed Abstract | CrossRef teljes szöveg | Google Scholar
14. Livadas S, Dracopoulou M, Dastamani A, Sertedaki A, Maniati-Christidi M, Magiakou A-M, et al. A klinikai, hormonális és molekuláris eredmények spektruma 280, a cyp21a2 gén mutációi által okozott, nem klasszikus veleszületett mellékvese hiperpláziában szenvedő egyénnél. Clin Endocrinol (Oxf). (2015) 82:543–9. doi: 10.1111 / cen.12543
PubMed Abstract | CrossRef teljes szöveg | Google Scholar
15. Bidé M, Bellanné-Chantelot C, Galand-Portier M-B, Tardy V, Billaud L, Laborde K, et al. Klinikai és molekuláris jellemzése egy kohorsz 161 független nők nem klasszikus veleszületett mellékvese hiperplázia miatt 21-hidroxiláz hiány és 330 családtag. J Clin Endocrinol Metab. (2009) 94:1570–8. doi: 10.1210 / jc.2008-1582
PubMed Abstract | CrossRef teljes szöveg | Google Scholar
16. Escobar-Morreale HF, Sanchón R, San Millán JL. A prospektív tanulmány a prevalenciája nem klasszikus veleszületett mellékvese hiperplázia a nők körében bemutató hiperandrogén tünetek, jelek. J Clin Endocrinol Metab. (2008) 93:527–33. doi: 10.1210 / jc.2007-2053
PubMed Abstract | CrossRef teljes szöveg | Google Scholar
17. Dewailly D, Vantyghem-Haudiquet MC, Sainsard C, Buvat J, Cappoen JP, Ardaens K, et al. Klinikai és biológiai fenotípusok a késői 21-hidroxiláz hiányban. J Clin Endocrinol Metab. (1986) 63:418–23. doi: 10.1210 / jcem-63-2-418
PubMed Abstract | CrossRef teljes szöveg | Google Scholar
18. Gönç EN, Ozön ZA, Alikaşifoglu A, Engiz O, Bulum B, Kandemir N. (2011). A basal szérum 17-OH progeszteron megbízható paraméter a nem klasszikus veleszületett mellékvese hiperplázia előrejelzésére a korai adrenarche-ban? Turk Jr. 53:274–80.
PubMed Abstract / Google Scholar
19. Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, et al. Veleszületett mellékvese hiperplázia szteroid 21-hidroxiláz hiány miatt: endokrin társadalom klinikai gyakorlat iránymutatás. J Clin Endocrinol Metab. (2010) 95:4133–60. doi: 10.1210 / jc.2009-2631
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Dacou-Voutetakis C, Dracopoulou M. High incidence of molecular defects of the CYP21 gene in patients with premature adrenarche. J Clin Endocrinol Metab. (1999) 84:1570–4. doi: 10.1210/jc.84.5.1570
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Ambroziak U, Kepczynska-Events I, Kurylowicz, Małunowicz em, Wojcicka, Miskiewicz P, et al. A mellékvesekéreg nemklasszikus veleszületett hiperpláziájának diagnosztizálása a 21-hidroxiláz hiány miatt, a szérum bazális vagy poszt-ACTH stimuláció 17-hidroxiprogeszteron alapján, hamis pozitív diagnózishoz vezethet. Clin Endocrinol (Oxf). (2016) 84:23–9. doi: 10.1111/ár 12935
PubMed Abstract / CrossRef teljes szöveg / Google Scholar
22. Ambroziak Y, Kepczynska-Events I, Kurylowicz, Wysłouch-Cieszyńska A, Małunowicz em, Bartoszewicz, et al. Az LC-MS / MS javítja a szűrést a 21-hidroxiláz hiány felé. Gynecol Endocrinol. (2015) 31:296–300. doi: 10.3109/09513590.2014.994599
PubMed Abstract | CrossRef teljes szöveg | Google Scholar
23. bbc, CC, Goldman MM, Zhang K, Clark-N, Reitz RE Welt CK. Tizenhárom szteroid hormon egyidejű mérése policisztás petefészek szindrómában szenvedő nőknél, valamint a folyadékkromatográfiás-tandem tömegspektrometriás kontroll nők ellenőrzése. PLoS One. (2014) 9:e93805. doi: 10.1371 / folyóirat.pone.0093805
PubMed Abstract | CrossRef teljes szöveg/Google Scholar
24. Ray JA, Kushnir MM, Yost RA, Rockwood AL, Wayne Meikle A. teljesítménynövelés az 5 endogén szteroidok mérésében LC-MS / MS differenciál ionmobilitási spektrometriával kombinálva. Clin Chim Acta. (2015) 438:330–6. doi: 10.1016 / j. cca.2014.07.036
PubMed Abstract | CrossRef teljes szöveg | Google Scholar
25. Stoupa A, González-Briceño L, Pinto G, Samara-Boustani D, Thalassinos C, Flechtner I, et al. Nem megfelelő kortizol válasz a tetracosactide (Synacthen®) tesztre nem klasszikus veleszületett mellékvese hiperpláziában: kivétel a szabály alól? Horm Res Gyermekgyógyász. (2015) 83:262–7. doi: 10.1159/000369901
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. (1992) 90:584–95. doi: 10.1172/JCI115897
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Temeck JW, Pang SY, Nelson C, New MI. A steroidogenezis genetikai hibái a korai pubarche-ban. J Clin Endocrinol Metab. (1987) 64:609–17. doi: 10.1210 / jcem-64-3-609
PubMed Abstract | CrossRef teljes szöveg | Google Scholar
28. Voutilainen R, Jääskeläinen J. korai adrenarche: etiológia, klinikai eredmények és következmények. J Szteroid Biochem Mol Biol. (2015) 145:226–36. doi: 10.1016 / j.jsbmb.2014.06.004
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Oberfield SE, Sopher AB, Gerken AT. Approach to the girl with early onset of pubic hair. J Clin Endocrinol Metab. (2011) 96:1610–22. doi: 10.1210/jc.2011-0225
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Janus D, Wojcik M, Malunowicz E, Starzyk JB. Ismétlődő labiális tapadások esetén egy 15 hónapos gyermeknél, tünetmentes, nem klasszikus veleszületett mellékvese hiperpláziával, a 21-hidroxiláz hiány miatt. J Pediatr Endokrinol Metab. (2012) 25:1017–21. doi: 10.1515/jpem-2012-0190
PubMed Abstract | CrossRef teljes szöveg/Google Scholar
31. Gopal-Kothandapani JS, Petkar A, O ‘ Shea E, Banerjee I. Perianal haj, mint a nem klasszikus veleszületett mellékvese hiperplázia szokatlan bemutatása. BMJ Case Rep. (2013) 2013: bcr2013010123. doi: 10.1136/bcr-2013-010123
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Falhammar H, Nordenström A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine. (2015) 50:32–50. doi: 10.1007/s12020-015-0656-0
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Chrousos GP, Loriaux DL, Mann DL, Cutler GB. Late-onset 21-hydroxylase deficiency mimicking idiopathic hirsutism or polycystic ovarian disease. Ann Intern Med. (1982) 96:143–8. doi: 10.7326/0003-4819-96-2-143
CrossRef Full Text | Google Scholar
34. Witchel SF. Non-classic congenital adrenal hyperplasia. Steroids. (2013) 78:747–50. doi: 10.1016/j.steroids.2013.04.010
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Carmina E, Rosato F, Jannì A, Rizzo M, Longo RA. Extensive clinical experience: relative prevalence of different androgen excess disorders in 950 women referred because of clinical hyperandrogenism. J Clin Endocrinol Metab. (2006) 91:2–6. doi: 10.1210/jc.2005-1457
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Moran C, Azziz R, Weintrob N, Witchel SF, Rohmer V, Dewailly D, et al. Reproduktív kimenetele nők 21-hidroxiláz-hiányos nemklasszikus mellékvese hiperplázia. J Clin Endocrinol Metab. (2006) 91:3451–6. doi: 10.1210 / jc.2006-0062
PubMed Abstract | CrossRef teljes szöveg | Google Scholar
37. Levin JH, Carmina E, Lobo RA. A policisztás petefészek szindrómában szenvedő betegek nem megfelelő gonadotropin szekréciója hasonló a felnőttkori veleszületett mellékvese hiperpláziában szenvedő betegekéhez? Fertil Steril. (1991) 56:635–40. doi: 10.1016/S0015-0282(16)54592-0
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Carmina E, Lobo RA. Ovarian suppression reduces clinical and endocrine expression of late-onset congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Fertil Steril. (1994) 62:738–43. doi: 10.1016/S0015-0282(16)56998-2
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Ludwig E. A női nemben előforduló androgenetikus alopecia (közös kopaszság) típusainak osztályozása. BRJ Dermatol. (1977) 97:247–54. doi: 10.1111 / j.1365-2133.1977.tb15179.x
PubMed Abstract | CrossRef teljes szöveg/Google Scholar
40. Trakakakis E, Papadavid e, Dalamaga M, Koumaki D, Stavrianeas N, Rigopoulos D, et al. Prevalenciája nem klasszikus veleszületett mellékvese hiperplázia miatt 21-hidroxiláz hiány görög nők akne: a kórházi keresztmetszeti vizsgálat. J Eur Acad Dermatol Venereol. (2013) 27:1448–51. doi: 10.1111 / j. 1468-3083. 2012. 04613.x
PubMed Abstract | CrossRef teljes szöveg/Google Scholar
41. Arlt W, Willis DS, Wild Sh, Krone N, Doherty EJ, Hahner S, et al. A veleszületett mellékvese hiperpláziában szenvedő felnőttek egészségi állapota: 203 beteg kohorsz vizsgálata. J Clin Endocrinol Metab. (2010) 95:5110–21. doi: 10.1210 / jc.2010-0917
PubMed Abstract | CrossRef teljes szöveg | Google Scholar
42. Saygili F, Oge A, Yilmaz C. Hyperinsulinemia és inzulinérzékenység a 21-hidroxiláz hiány miatt nem klasszikus veleszületett mellékvese hiperpláziában szenvedő nőknél: a szérum leptinszint és a krónikus hyperinsulinaemia közötti kapcsolat. Horm Res (2005) 63:270-4. doi: 10.1159/000086363
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Speiser PW, Serrat J, New MI, Gertner JM. Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1992) 75:1421–4. doi: 10.1210/jc.75.6.1421
PubMed Abstract | CrossRef Full Text | Google Scholar
44. Han TS, Stimson RH, Rees DA, Krone N, Willis DS, Conway GS, et al. Glükokortikoid kezelés és egészségügyi eredmények veleszületett mellékvese hiperpláziában szenvedő felnőtteknél. Clin Endocrinol. (2013) 78:197–203. doi: 10.1111 / cen.12045
PubMed Abstract | CrossRef teljes szöveg | Google Scholar
45. Van Staa TP, LEUFKENS HG, Abenhaim L, Zhang B, Cooper C. orális kortikoszteroidok alkalmazása és a törések kockázata. J Bone Miner Res. (2000) 15:993-1000. doi: 10.1359 / jbmr.2000.15.6.993
PubMed Abstract | CrossRef Full Text | Google Scholar
46. Nebesio TD, Renbarger JL, Nabhan ZM, Ross SE, Slaven JE, Li L, et al. Differential effects of hydrocortisone, prednisone, and dexamethasone on hormonal and pharmacokinetic profiles: a pilot study in children with congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2016) 2016:17. doi: 10.1186 / s13633-016-0035-5
PubMed Abstract | CrossRef teljes szöveg | Google Scholar
47. Weintrob N, Dickerman Z, Sprecher E, Galatzer A, Pertzelan A. Nem klasszikus 21-hidroxiláz hiány csecsemőkorban és gyermekkorban: a kezelés megkezdésének ideje hatása a pubertásra és a végső magasságra. Eur J Endocrinol. (1997) 136:188–95. doi: 10.1530 / eje.0.1360188
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Bonfig W, Bechtold S, Schmidt H, Knorr D, Schwarz HP. Reduced final height outcome in congenital adrenal hyperplasia under prednisone treatment: deceleration of growth velocity during puberty. J Clin Endocrinol Metab. (2007) 92:1635–9. doi: 10.1210/jc.2006-2109
PubMed Abstract | CrossRef Full Text | Google Scholar
49. Dörr HG. Növekedés a klasszikus veleszületett mellékvese hiperpláziában szenvedő betegeknél a 21-hidroxiláz hiány miatt. Horm Res (2007) 68(5.): 93-9. doi: 10.1159/000110587
PubMed Abstract / CrossRef teljes szöveg / Google Scholar
50. Hoepffner W, Kaufhold A, Willgerodt H, Keller E. A klasszikus veleszületett mellékvese hiperpláziában szenvedő betegek a 21-hidroxiláz hiány miatt elérhetik célmagasságukat: a Lipcsei tapasztalat. Horm Res (2008) 70:42-50. doi: 10.1159/000129677
PubMed Abstract | CrossRef Full Text | Google Scholar
51. van der Kaay D, van den Akker E. Ultralow-dose dexamethasone to preserve endogenous cortisol stress response in nonclassical congenital adrenal hyperplasia: a new promising treatment. Int J Endocrinol Metab. (2014) 12:e14657. doi: 10.5812/ijem.14657
CrossRef Full Text | Google Scholar
52. Lin-Su K, Vogiatzi MG, Marshall I, Harbison MD, Macapagal MC, Betensky B, et al. Treatment with growth hormone and luteinizing hormone releasing hormone analog improves final adult height in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. (2005) 90:3318–25. doi: 10.1210/jc.2004-2128
PubMed Abstract | CrossRef Full Text | Google Scholar
53. McCann-Crosby B, Chen M-J, Lyons SK, Lin Y, Axelrad M, Dietrich JE, et al. Nonclassical congenital adrenal hyperplasia: a kezelés és az átmenet céljai. Pediatr Endokrinol Rev. (2014) 12: 224-38.
PubMed Abstract | Google Scholar
54. Ogilvie CM, Crouch NS, Rumsby G, Creighton SM, Liao L-M, Conway GS. Veleszületett mellékvese hiperplázia felnőtteknél: orvosi, sebészeti, pszichológiai kérdések áttekintése. Clin Endocrinol. (2006) 64:2–11. doi: 10.1111 / j. 1365-2265.2005. 02410.x
PubMed Abstract | CrossRef Full Text | Google Scholar
55. Mah PM, Jenkins RC, Rostami-Hodjegan A, Newell-Price J, Doane A, Ibbotson V, et al. Weight-related dosing, timing and monitoring hydrocortisone replacement therapy in patients with adrenal insufficiency. Clin Endocrinol. (2004) 61:367–75. doi: 10.1111/j.1365-2265.2004.02106.x
PubMed Abstract | CrossRef Full Text | Google Scholar
56. Diaz A, Laufer Mr Breech LLAA PC on A, Care AC of O és GC on AH. Menstruáció lányoknál és serdülőknél: a menstruációs ciklus használata létfontosságú jel. Gyermekgyógyászat. (2006) 118:2245–50. doi: 10.1542 / peds.2006-2481
CrossRef teljes szöveg | Google Scholar
57. Merke DP, Poppas DP. A veleszületett mellékvese hiperpláziában szenvedő serdülők kezelése. Lancet Diabetes Endocrinol. (2013) 1:341–52. doi: 10.1016/S2213-8587(13)70138-4
PubMed Abstract | CrossRef Full Text | Google Scholar
58. Verma S, Green-Golan L, VanRyzin C, Drinkard B, Mehta SP, Weise M, et al. Adrenomedullary function in patients with nonclassic congenital adrenal hyperplasia. Horm Metab Res. (2010) 42:607–12. doi: 10.1055/s-0030-1253385
PubMed Abstract | CrossRef Full Text | Google Scholar
59. New MI. Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2006) 91:4205–14. doi: 10.1210/jc.2006-1645
PubMed Abstract | CrossRef Full Text | Google Scholar
60. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2018) 103:4043–88. doi: 10.1210/jc.2018-01865
PubMed Abstract | CrossRef Full Text | Google Scholar
61. Kruse B, Riepe FG, Krone N, Bosinski HAG, Kloehn S, Partsch CJ, et al. Congenital adrenal hyperplasia – how to improve the transition from adolescence to adult life. Exp Clin Endocrinol Diabetes. (2004) 112:343–55. doi: 10.1055/s-2004-821013
PubMed Abstract | CrossRef Full Text | Google Scholar
62. Gastaud F, Bouvattier C, Duranteau L, Brauner R, Thibaud E, Kutten F, et al. Károsodott szexuális és reproduktív eredmények a veleszületett mellékvese hiperplázia klasszikus formáival rendelkező nőknél. J Clin Endocrinol Metab. (2007) 92:1391–6. doi: 10.1210 / jc.2006-1757
PubMed Abstract | CrossRef teljes szöveg | Google Scholar
63. Mercè Fernández-Balsells M, Muthusamy K, Smushkin G, Lampropulos JF, ELAMIN MB, Abu Elnour NO, et al. Prenatális dexametazon alkalmazása a klasszikus veleszületett mellékvese hiperplázia kockázatának kitett terhességek virilizációjának megelőzésére 21-hidroxiláz (CYP21A2) hiány miatt: szisztematikus felülvizsgálat és meta-analízis. Clin Endocrinol. (2010) 73:436–44. doi: 10.1111 / j. 1365-2265. 2010. 03826.x
CrossRef teljes szöveg/Google Scholar
64. Bidé M, Bellanné-Chantelot C, Galand-Portier MB, Golmard JL, Tardy V, Morel Y, et al. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2010) 95:1182–90. doi: 10.1210/jc.2009-1383
PubMed Abstract | CrossRef Full Text | Google Scholar
65. Birnbaum MD, Rose LI. Late onset adrenocortical hydroxylase deficiencies associated with menstrual dysfunction. Obstet Gynecol. (1984) 63:445–51.
PubMed Abstract | Google Scholar
66. Cuhaci N, Aydin C, Yesilyurt A, Pinarli FA, Ersoy R, Cakir B. Nonclassical congenital adrenal hyperplasia and pregnancy. Case Rep Endocrinol. (2015) 2015:296924. doi: 10.1155/2015/296924
PubMed Abstract | CrossRef Full Text | Google Scholar
67. Deneux C, Tardy V, Anne D, Mornet E, Billaud L, Charron D, et al. Fenotípus-genotípus korreláció 56 olyan nőben, akiknek nem klasszikus veleszületett mellékvese hiperpláziája van a 21-hidroxiláz hiány miatt. J Clin Endocrinol Metab. (2001) 86:207–13. doi: 10.1210 / jcem.86.1.7131
PubMed Abstract | CrossRef teljes szöveg | Google Scholar
68. Simonetti L, Bruque CD, Fernández CS, Benavides-Mori B, Delea M, Kolomenski JE, et al. Cyp21a2 mutáció frissítés: adatbázisok átfogó elemzése és közzétett genetikai változatok. Hum Mutat. (2018) 39:5–22. doi: 10.1002 / humu.23351
PubMed Abstract | CrossRef Full Text | Google Scholar
69. OMIM Entry – * 613815 – CYTOCHROME P450 FAMILY 21 SUBFAMILY A POLYPEPTIDE 2; CYP21A2.
Google Scholar
70. Miller WL, Witchel SF. Prenatal treatment of congenital adrenal hyperplasia: risks outweigh benefits. Am J Obstet Gynecol. (2013) 208:354–9. doi: 10.1016/j.ajog.2012.10.885
PubMed Abstract | CrossRef Full Text | Google Scholar
71. Lekarev O, New MI. Adrenal disease in pregnancy. Best Pract Res Clin Endocrinol Metab. (2011) 25:959–73. doi: 10.1016/j.beem.2011.08.004
PubMed Abstract | CrossRef Full Text | Google Scholar
72. New MI, Abraham M, Yuen T, Lekarev O. An update on prenatal diagnosis and treatment of congenital adrenal hyperplasia. Semin Reprod Med. (2012) 30:396–9. doi: 10.1055/s-0032-1324723
PubMed Abstract | CrossRef Full Text | Google Scholar
73. Kazmi D, Bailey J, Yau M, Abu-Amer W, Kumar A, Low M, et al. New developments in prenatal diagnosis of congenital adrenal hyperplasia. J Steroid Biochem Mol Biol. (2017) 165:121–3. doi: 10.1016/j.jsbmb.2016.06.016
PubMed Abstract | CrossRef Full Text | Google Scholar
74. Clayton PE, Miller WL, Oberfield SE, Ritzén EM, Sippell WG, Speiser PW, et al. Konszenzus nyilatkozat Az Európai Gyermekgyógyászati Endokrinológiai Társaság és a lawson Wilkins gyermekgyógyászati Endokrin Társaság 21-hidroxiláz hiányáról. Horm Res (2002) 58:188-95. doi: 10.1159/000065490
PubMed Abstract / CrossRef teljes szöveg / Google Scholar
75. Nandagopal R, Sinaii N, Avila NA, Van Ryzin C, Chen W, Finkielstain GP, et al. Fenotípusos profilalkotás a szülők rejtélyes nemklasszikus veleszületett mellékvese hiperplázia: megállapítások 145 független családok. Eur J Endocrinol. (2011) 164:977–84. doi: 10.1530/EJE-11-0019
PubMed Abstract | CrossRef teljes szöveg/Google Scholar
76. Takasu N, Nakachi K, Higa H. A Graves hyperthyreosisának kialakulása mellékvese válságot okozott egy korábban nem ismert, nem klasszikus 21-hidroxiláz-hiányban szenvedő betegben. Gyakornok Orvos. (2010) 49:1395–400. doi: 10.2169 / internalmedicine.49.3573
PubMed Abstract | CrossRef Full Text | Google Scholar
77. Meyer-Bahlburg HFL, Dolezal C, Baker SW, Carlson AD, Obeid JS, New MI. Prenatal androgenization affects gender-related behavior but not gender identity in 5-12-year-old girls with congenital adrenal hyperplasia. Arch Sex Behav. (2004) 33:97–104. doi: 10.1023/B:ASEB.0000014324.25718.51
CrossRef Full Text | Google Scholar
78. Meyer-Bahlburg HFL, Dolezal C, Baker SW, New MI. Szexuális irányultság a klasszikus vagy nem klasszikus veleszületett mellékvese hiperpláziában szenvedő nőknél a prenatális androgén felesleg mértékének függvényében. Arch Szex Viselkedik. (2008) 37:85–99. doi: 10.1007 / s10508-007-9265-1
PubMed Abstract / CrossRef teljes szöveg / Google Scholar