- definitioner och prevalens
- diagnos
- Nccah manifestationer hos kvinnor
- manifestationer i barndomen
- manifestationer i tonåren och vuxenlivet
- Nccah-hantering från den första manifestationen till vuxenlivet
- hantering under barndomen
- hantering under tonåren
- patienter som behandlats sedan barndomen
- kvinnor med första Manifestation under tonåren eller hyperandrogena symtom efter avbrytande av behandlingen
- NCCAH och reproduktion
- subfertilitet
- missfall
- Fertilitetsplanering
- Prenatal behandling av CAH
- behandling under graviditet
- andra speciella problem
- stresshantering i NCCAH
- psykobiologiska aspekter i NCCAH
- slutsatser
- Författarbidrag
- intressekonflikt uttalande
definitioner och prevalens
medfödd adrenal hyperplasi (CAH) omfattar en familj av autosomala recessiva störningar som kännetecknas av mild till akut nedsatt kortisolsyntes på grund av brist i en av de fem adrenal steroidogena enzymerna som krävs för kortisolproduktion (1, 2). Konventionellt är CAH uppdelad i (A) klassisk (CCAH), presenterar med saltavfall eller den enkla viriliserande formen som manifesteras vid födseln och/eller i neonatalperioden, och (b) icke-klassisk (NCCAH), som representerar en mindre allvarlig form av sjukdomen som saknar genital tvetydighet, är inte omedelbart livshotande och presenterar senare i livet, eller förblir asymptomatisk eller är feldiagnostiserad som en annan sjukdom (3). Gränserna över de olika formerna av CAH är dock inte strikt definierade, vilket tenderar att öka utmaningarna i samband med denna störning. Det är därför lämpligt att överväga CAH som ett kontinuum av fenotyper, från svår till mild eller annars asymptomatisk (3).
den vanligaste formen av CAH orsakas av 21-hydroxylasbrist (21ohd), vilket resulterar i försämrad eller ingen omvandling av 17 hydroxiprogesteron (17 OHP) till 11 deoxikortisol och progesteron till deoxikortikosteron. Blockaden av steroidkonvertering resulterar i ökad produktion av androgenprekursorer, under CRH-ACTH-stimulering, vilket leder till biokemisk hyperandrogenism, markerad av förhöjda 17-OHP-nivåer. Den klassiska formen av sjukdomen förekommer hos 1 av 16 000 levande födda över hela världen. Det bör dock nämnas att den upplevda förekomsten av sjukdomen är relaterad till den använda screeningmetoden. Det har till exempel rapporterats att förekomsten av den klassiska formen av CAH i Sverige är 1:11 500 när en fallundersökningsmetod används, vilken siffra sjunker dock till 1:9 800 när hormonell screening tillämpas. På samma sätt varierar incidensen i Frankrike och Schweiz mellan 1:15,472 och 1:23,000 och 1:10,749 och 1:11,661 när de olika metoderna för screening används (4).
NCCAH är mycket vanligare, förekommer hos cirka 1 av 1000 kaukasier och oftare i vissa etniska grupper, såsom Ashkenazi-judar (1: 27), latinamerikaner( 1: 53), jugoslaver (1: 62) och italienare (1:300) (5, 6). Ändå saknas data om förekomsten av NCCAH baserat på olika uppskattningsmetoder (fallundersökning, hormonell screening och molekylär testning). NCCAH anses vara den vanligaste autosomala recessiva endokrina störningen med en bärarfrekvens på 1:25 till 1:10. I NCCAH på grund av 21-OHD uppskattas den återstående enzymatiska aktiviteten vara cirka 10-70% (7-9).
diagnos
i jämförelse med diagnosen av den klassiska formen av sjukdomen, som görs vid födseln eller under neonatalperioden på grund av genital tvetydighet och/eller saltavfallssymptom eller genom screeningprogram som används i vissa länder är de flesta fall av NCCAH inte lätt detekterbara (4, 10). Dessutom förblir många individer asymptomatiska under barndomen och tonåren, har normal reproduktiv funktion och blir bara medvetna om NCCAH på grund av diagnosen av en annan familjemedlem och därmed testning (11). De flesta kvinnor med NCCAH söker dock medicinsk hjälp när de upplever symtom på androgenöverskott och, när klinisk misstanke uppmanar testning, förhöjda basala 17 OHP-nivåer kommer sannolikt än inte att peka på en diagnos av NCCAH.
faktum är att de kliniska riktlinjerna som föreslagits av Endocrine Society rekommenderar ett icke-stimulerat värde på 17 OHP som screeningverktyg för NCCAH. Morgon 17 OHP nivåer > 6 nmol/L i follikulärfasen hos menstruerande kvinnor bör leda till ytterligare utvärdering, eftersom det har visats att nivåer över 6 nmol/L fångar 90% av nccah-individer (12, 13). Slumpmässiga mätningar av 17 OHP har inte visat sig vara till hjälp, eftersom dessa ofta ger normala nivåer hos patienter med NCCAH, och de är dessutom extremt höga i lutealfasen hos hälften av friska kvinnor (13). Men i våra data härledda från 280 personer med sjukdomen, sex patienter (2.1%) hade ett utgångsvärde på 17 OHP < 6 nmol/L (14). Dessutom, Bidet et al., i en stor kohort av kvinnor med NCCAH verifierad med molekylära tekniker fann också basala 17 OHP-värden lägre än 6 nmol/L hos 8% av de studerade ämnena (15). Slutligen, baserat på data som samlats in av Speiser et al., 9% av individerna med NCCAH visade 17 OHP-värden lägre än 2 ng/ml (det motsvarar 6nmol/L). Enligt andra studier är ett baslinjevärde på 17 OHP mellan 5, 1 och 9 nmol/L tillräckligt för diagnos av NCCAH (13, 16, 17). Nyligen en nivå av basal 17 OHP av 4.6 nmol / L föreslogs som en tröskel för ACTH-testning för att förutsäga NCCAH hos patienter med för tidig adrenarche under barndomen (18).
summan av dessa fynd och förslag indikerar att valet av patienter som kommer att genomgå ett synacthen-stimuleringstest bör utvärderas från fall till fall. En 17 OHP efter stimuleringsnivå över 3 nmol/L krävs för diagnosen (19). Heterozygoter som bär en cyp21a2-mutation uppvisar något förhöjda 17 OHP-nivåer efter ACTH-stimulering, även om det finns överlappning hos opåverkade personer (9). Dock, som Dacou-Voutetakis et al. har påpekat, om summan av basala och poststimulerande 17 OHP-värden överstiger 1,5 nmol/L, är möjligheten till heterozygositet exceptionellt hög (20).
en annan viktig faktor gäller de tekniker som används för 17 OHP utvärdering. 17 OHP-nivåer mäts med olika immunanalysmetoder, men som nyligen visats uppnåddes de mest exakta och tillförlitliga resultaten genom implementeringen av kombinationen av vätskekromatografi med masspektrometri (LC-MS/MS). Faktum är att många falska positiva 17 OHP-mätningar hittades när LC-MS/Ms-mätningar jämfördes med standardmetoder (21) ändå används de senare procedurerna inte allmänt, i händelse av en osäker diagnos kommer de att ge mer exakta resultat. Observera att tröskelvärdena för screening och diagnostik för LC-MS/MS kvantifierad 17 OHP ännu inte har definierats. Dessa tröskelvärden är sannolikt lägre än de som fastställts med immunanalyser på grund av den förbättrade specificiteten hos LC-MS/MS, en metod som är mindre benägen för korsreaktivitet och störningar (22, 23). Huruvida en urinsteroid profil krävs för den definitiva diagnosen återstår att klargöras (22). I gränsfall är det lämpligt att erhålla en fullständig adrenokortisk profil efter ACTH-stimuleringstestet för att skilja 21-hydroxylasbrist från andra enzymdefekter och upprätta en fast diagnos.
specifikt bör en fullständig steroidprofil utföras i tvetydiga fall för att bekräfta 21-hydroxylasbrist och utesluta andra enzymatiska detekteringar. Inkludering av 17 OHP, kortisol, 11-deoxikortikosteron,11-deoxikortisol,17-OH-pregnenolon, Dehydroepiandrosteron och androstenedion i en serumsteroidprofil skulle vara användbar för att utesluta andra orsaker till CAH, såsom 11 kg-hydroxylasbrist och, mer sällan, 3 kg-hydroxisteroiddehydrogenasbrist eller P450 oxidoreduktasbrist (24). Även om genetisk testning inte anses vara ett primärt diagnostiskt verktyg för NCCAH vid denna tidpunkt är det obligatoriskt för diagnosbekräftelse och för genetisk rådgivning (3).
När det gäller kortisolnivåerna förväntas i allmänhet ett poststimuleringsvärde på minst 496 nmol/L. Observera, enligt Stoupa et al., 60% av 47 barn med NCCAH som ett resultat av 21 OHD hade låga kortisolvärden efter stimuleringen, ett konstaterande som pekade på behovet av ökad övervakning för utveckling av binjurinsufficiens under stora stresshändelser (25).
Nccah manifestationer hos kvinnor
det kliniska uttrycket av NCCAH kännetecknas av en hög nivå av polymorfism när det gäller inte bara ålder av början utan också de olika tecknen och symtomen. Det rapporteras att den första kliniska presentationen av NCCAH är i 11% av fallen före 10 års ålder och i 80% mellan 10 och 40 år (12). Korrelationen mellan genotyp och fenotyp I CAH och NCCAH har ännu inte klarlagts. Speiser et al. föreslå att de flesta men inte alla fenotypiska variationerna i 21-hydroxylasbrist beror på allelvariation i CYP21A2 (26).
manifestationer i barndomen
i den nyfödda perioden förblir vanligtvis NCCAH-kvinnor asymptomatiska och har normala yttre könsorgan. Det tidigaste fallet med nccah rapporterade är en 6 månader gammal tjej som utvecklade könshår (11). Vanligtvis börjar kliniska fynd och symtom i nccah-fall från 5 års ålder eller till och med senare och är relaterade till ökade androgennivåer, men mild kortisolbrist kan också förekomma i vissa fall (8). I 92% av nccah-fallen är det första symptomet som uppträder för tidigt pubarche (PP), som förekommer under 8 år hos flickor och under 9 år hos Pojkar (12) med en uppskattad prevalens på mellan 10 och 11, 3% (15). Observera att hos barn som hänvisas till PP, enligt olika studier, varierar förekomsten av NCCAH från 5 till 30% (20, 27). Baserat på vår analys av 280 patienter med molekylär bekräftelse av NCCAH fann vi dock att förekomsten av PP hos 94 kvinnor yngre än 8 år var så mycket som 88%. Å andra sidan identifierade en analys av 45 män med NCCAH PP hos endast 29% av patienterna (14). Dessutom bör vi komma ihåg att ungefär hälften av patienterna med PP kan vara heterozygotbärare av en CYP21A2-mutation.
en annan aspekt som behöver förtydligas är förhållandet mellan PP och tidig pubertet. Det antas att i vissa fall kan perifer aromatisering av adrenal androgener till östrogener aktivera hypotalamus-hypofys-gonadalaxeln, vilket leder till sekundär tidig pubertet (28). I ett sådant fall behövs regelbunden uppföljning. Vissa barn utvecklar hirsutism, akne och/eller snabb tillväxt och uppenbarar hög struktur och benålders framsteg (29). Den senare kan resultera i trunkerad slutlig höjd som en följd av snabb epifyseal fusion. Andra sällsynta kliniska fynd under barndomen inkluderar labial vidhäftning, perianalt hår, klitoromegali och heshet av röst (30-32).
manifestationer i tonåren och vuxenlivet
under tonåren och vuxenlivet presenterar NCCAH hos kvinnor hyperandrogena symtom som liknar polycystiskt äggstockssyndrom (PCOS) (33). Symtom inkluderar hirsutism, akne, androgen alopeci, anovulation, menstruationsdysfunktion och infertilitet. I en multicenterstudie var de vanligaste symtomen bland ungdomar och vuxna kvinnor hirsutism (59%), oligomenorrhea (54%) och akne (33%). Bland 161 kvinnor med NCCAH var symtom hirsutism (78%), menstruationsdysfunktion (54,7%) och minskad fertilitet (12%) (34). I vår grupp av 161 kvinnor med sjukdomen 63% av patienten presenteras med en polycystisk äggstocksliknande fenotyp (14). Detta resultat har en vice versa implikation, eftersom förekomsten av NCCAH är hög hos kvinnor med hyperandrogenemi. Visst, studien av Witcel et al. att rapportera en förekomst av NCCAH som sträcker sig från 1 till 33% hos hyperandrogena kvinnor är av stort intresse. I en annan studie av 950 kvinnor som hänvisades till utvärdering av androgenöverskott upptäcktes NCCAH hos 4,2% av dem. Nccah-patienterna var dock yngre och mer hirsute jämfört med de andra grupperna av hyperandrogena patienter. De kännetecknades också av signifikant högre nivåer av testosteron, fritt testosteron och 17 OHP än patienter med andra hyperandrogena syndrom. Vid ultraljud i äggstockarna visade 77% av dem polycystiska äggstockar och 41% ökade äggstocksstorleken. Sammanfattningsvis uppfyllde de PCOS-kriterierna enligt NIH och Rotterdam med en procentandel av 56 respektive 72,8% (35).
sjukdomens progressiva natur framhävs av det faktum att förekomsten av hirsutism har visat sig öka med åldern och har observerats vara sällsynt före puberteten. Hirsutism är den vanligaste kliniska manifestationen som rapporterats hos patienter med NCCAH, från 71 till 96% (28, 36). Pubertal flickor med NCCAH vanligtvis närvarande med hirsutism (37, 38). I den andra änden av spektrumet är frågan om alopeci. Manligt skallighet rapporteras hos nccah-patienter. Förekomsten av alopeci verkade också öka med åldern, från 6% hos patienter under det andra decenniet av sitt liv till 19% i det femte, vilket återigen indikerar sjukdomens progressiva natur (39). Akne rapporteras hos nästan 33% av nccah-patienterna (36). Anmärkningsvärt upptäcktes mutationer av CYP21A2-genen som orsakade NCCAH hos 4,9% av 123 vuxna kvinnor med svår akne (40).
menstruella oegentligheter inklusive oligomenorrhea eller sekundär amenorrhea kan ofta vara det presenterande tecknet på NCCAH hos post menarche individer. Dessutom upplever 8-9% av fallen också primär amenorrhea och söker därför läkare för första gången. Dessutom, bland nccah individer, oligomenorrhea är vanligare än primär amenorrhea. Till exempel i en serie från Moran et al., bland NCCAH-kvinnor i åldern 10 till 19 år var oligomenorrhea närvarande i 56% av fallen jämfört med endast 9% av tonåren med primär amenorrhea (12). Sammanfattningsvis liknar så många nccah-symtom PCOS att det inte är förvånande att NCCAH har kallats PCOS stora mimicker. I vilket fall som helst bör en diagnos av NCCAH övervägas vid utvärderingen av en ung kvinna som hänvisas till hyperandrogena symtom.
i en studie av Arlt et al., patienter med NCCAH kännetecknades av signifikant högre BMI och lägre sluthöjd jämfört med matchade kontroller (41). Dessa data, i kombination med några studier som indikerar närvaron av insulinkänslighet, föreslår en ogynnsam metabolisk profil som kan patofysiologiskt förklaras av effekten av förhöjda androgenkoncentrationer och/eller som ett resultat av glukokortikoidbehandling (42, 43). Osteoporos kan också detekteras hos dessa personer, troligen som en följd av kortikosteroidterapin (34).
Nccah-hantering från den första manifestationen till vuxenlivet
När diagnosen NCCAH har fastställts, motiverar flera frågor relaterade till efterföljande behandlingsrekommendationer övervägande. Den första frågan som ska behandlas är om glukokortikoidbehandling (GC) indikeras. Den allmänna resonemanget bakom detta tillvägagångssätt är att genom att tillhandahålla tillräckliga kortisolkoncentrationer till patienten täcks hennes dagliga behov och följaktligen kommer CRH-ACTH-axelstimulering att avsmalna, vilket leder till minskad adrenal androgenproduktion. Den allmänna regeln om detta tillvägagångssätt är att GCs ges vid ersättning och inte farmakologiska doser, medan påverkan av ålder, kön, laboratoriedata, patientspecifika rekommendationer och mål för behandling av glukokortikoidersättningsterapi beaktas allvarligt. Vi bör dock också vara medvetna om att långvarig GC-substitutionsbehandling kan leda till hyperkortisolemi, med de resulterande väldokumenterade negativa effekterna på alla aspekter av ämnesomsättningen, särskilt tillväxt, fettfördelning och insulinresistens samt på psykologisk profil. En annan stor nackdel med detta tillvägagångssätt är bristen på ett adekvat kliniskt index eller biokemisk markör för adekvat ersättningsdosering, såsom existerar angående TSH-värden vid hypotyroidism.slutligen komplicerar det faktum att det inte finns någon konsensus om vilken GC som ska användas och frånvaron av långsiktiga data om olika metoder för GC-administrering ytterligare behandlande läkares beslut om och val av det optimala valet för varje patient. Hydrokortison används vanligtvis hos barn, eftersom det mest liknar det naturliga hormonet (kortisol), men det anses inte vara ett lämpligt tillvägagångssätt hos ungdomar och unga kvinnor på grund av behovet av upprepad daglig dosering. Därför föredrar de flesta vuxna endokrinologer antingen dexametason eller Prednisolon, vid lämpliga doser. Å andra sidan måste det påpekas att ekvivalensen av olika GCs baseras på deras antiinflammatoriska verkan och inte på olika aspekter av mänsklig metabolism. Eftersom kortisol påverkar nästan 20% av det mänskliga genomet, förväntas olika svar från olika GCs i olika vävnader. Att bekräfta denna uppfattning är det faktum att administrering av dexametason till patienter med CAH har visat försämring av index för insulinresistens jämfört med andra GCs (44). Dessutom har administreringen av 2,5-7,5 mg prednisolon, en dos som anses vara normal, en långvarig negativ inverkan på benmetabolismen (45). Därför bör hydrokortison anses vara den bästa behandlingsformen vid GC-tilläggsbehandling. Tillkomsten av den nyligen syntetiserade hydrokortisonformeln med ett piller per dag och dess initiala positiva resultat hos patienter med CAH visar mycket löfte för framtiden (46).
hantering under barndomen
nccah-patienter som diagnostiseras under barndomen med tecken på PP kan behandlas med hydrokortison i syfte att undertrycka binjurhormonerna och förhindra snabb utveckling av benåldern som kan påverka slutlig höjd. Hos de patienter hos vilka hydrokortisonbehandling inleddes 1 år före puberteten och som hade en benålder under 9 år förblev den slutliga höjden inom den genetiska potentialen (47). Tillgängliga studier tyder på att vuxenhöjd närmade sig den förväntade målhöjden hos patienter som övervakades noggrant och som strikt följde medicineringsplaner (48-50). En ny liten studie av fem fall (patienter 6, 1-9, 2 år) visade att en ultralåg dos dexametason är ett lovande alternativ för att minska endogen stress och dess effekter. Huruvida detta är ett realistiskt tillvägagångssätt och hur mycket den administrerade dosen ska vara återstår att klarlägga (51). En studie av Nebesio et al. jämförde de hormonella effekterna och farmakokinetiken hos hydrokortison, prednisolon och dexametason hos 9 prepubertala patienter med CCAH. De visade att dexametason var mer potent att de andra formerna för att uppnå signifikant lägre binjurhormonnivåer, vilket tyder på att dexametason är mer effektivt för undertryckande av adrenal androgenproduktion (46). Naturligtvis behövs ytterligare studier för att verifiera eller avvisa detta resultat. Dessutom kan förhöjda androgenkoncentrationer i vissa fall leda till sekundär stimulering av GnRH-axeln, vilket leder till för tidig pubertet. I ett sådant fall kan en parallell kurs med GnRH-analog förskrivas om benåldern är betydligt högre än kronologisk ålder och/eller projicerad sluthöjd är oproportionerlig mot målhöjd.
ett annat viktigt övervägande gäller fall av samtidig GH-brist i händelse av att behandling med GnRH-analoger och GH-recept resulterade i att målhöjden uppnåddes (52). Enligt de nuvarande riktlinjerna rekommenderas den ovan beskrivna behandlingen försiktigt endast för de fall där den förutsagda höjden SD är -2,25 < målhöjden eller till och med lägre (53). En alternativ indikation för att starta hydrokortisonbehandling är ett otillräckligt kortisolrespons efter ACTH-stimulering (36).den föredragna GC-behandlingen hos barn är vanligtvis hydrokortison 10-15 mg / m2, uppdelad i tre doser. Ofta har lägre doser också visat sig vara effektiva, från 6 mg/m2 / dag (34). Överdosering bör undvikas, med tanke på det som kan leda till dålig tillväxt samt Cushingoid funktioner. Regelbundet tillväxtmönster, en benålder som är kompatibel med kronologisk ålder och frånvaro av central fetma kan också fungera som kliniska index för lämplig hantering. När det gäller biokemisk/ hormonell profil anses androstenedion-och testosteronnivåer i mitten till övre intervall för benålder vara bättre markörer för adekvat GC-ersättningsterapi hos barn med NCCAH. Undertryckta testosteronnivåer hittades emellertid hos 10% av nccah-patienterna, medan ytterligare 28% av patienterna hade ökat testosteronkoncentrationer, vilket möjligen kan hänföras till hydrokortisonvariabilitet (54, 55). DHEAS-värdena minskas med mycket små doser, medan undertryckandet av 17 OHP och progesteronnivåer kräver mycket höga GC-doser. Det bör noteras att 17 OHP–värden är avgörande för diagnosen, men inte till hjälp vid uppföljning. Observera att blodprovtagning för hormonell utvärdering måste utföras utan att behandlingen avslutas. På grund av sjukdomens förvirring och dess mångfacetterade natur finns det emellertid inga specifika riktlinjer för tidpunkten för regimförändringar eller upphörande av glukokortikoidbehandling hos barn.
hantering under tonåren
patienter som behandlats sedan barndomen
tills etableringen av det normala menstruationsmönstret hos NCCAH-tjejer rekommenderas starkt fortsättningen av GCs som startade under barndomen (56). Ungdomspatienter visar emellertid ofta inte tillräcklig överensstämmelse med kronisk administrering av läkemedel och utelämnar ofta doser. Dessutom, under puberteten, halveringstiden för hydrokortison sjunker med 50% som ett resultat av ökade IGF-1 nivåer, vilket minskar 11 ci-ohsd aktivitet, liksom på grund av ökad kortisol clearance härrör från förstärkning av glomerulär filtreringshastighet (57). Hos den unga kvinnan, 2 år efter menarche och om normala ägglossningscykler har registrerats, rekommenderas starkt ett patientcentrerat tillvägagångssätt mot de hyperandrogena symtomen som kan uppstå.
om det finns allvarliga hyperandrogena fynd, såsom hirsutism, akne och/eller oligomenorrhea, kommer fortsatt behandling att övervägas. Det är viktigt att komma ihåg de bräckliga känslorna hos en ung tonåring, som kommer att skadas allvarligt av de ”avstötande” tecknen på hyperandrogenism. Kombinationen av hyperandrogena tecken, menstruationsstörningar och dålig livskvalitet är väl dokumenterad och den är särskilt hög hos yngre kvinnor. En annan punkt som ska beaktas är svaret på adrenal reserve post ACTH stimulering. Med tanke på att toppkortisol hos pubertala och vuxna kvinnor efter stimulering är under 496 nmol/L, i fall av stress, bör steroidbehandling administreras (58). Å andra sidan, om inget av ovanstående symtom uppträder hos den unga patienten, kan GC-behandlingen avbrytas, varefter regelbunden uppföljning rekommenderas.
kvinnor med första Manifestation under tonåren eller hyperandrogena symtom efter avbrytande av behandlingen
i närvaro av hyperandrogena symtom rekommenderas ett patientorienterat tillvägagångssätt med fokus på patientens huvudklagomål. Detta är mycket bättre än en allmän universell modus operandi, eftersom den unga patienten med den senare metoden kommer att bli besviken, vilket leder till att behandlingen avbryts och som ett resultat till negativa hälsoutfall i vuxen ålder. I fall med svår hirsutism utgör en kurs på 6 till 12 månader med p-piller (OCPs) ett rimligt förfarande med tanke på de positiva effekterna av OCPs på SHBG-leverproduktionen och minskningen av androgenfrisättning från äggstocken. Klinisk förbättring förväntas efter minst 2-3 månaders OCPs-initiering, medan samtidig användning av ett progestagen med minimala androgena egenskaper rekommenderas starkt. Om OCP misslyckas som första linjens tillvägagångssätt kan antiandrogener (spironolakton, Flutamid, bikalutamid, cyproteronacetat och finasterid) tillsättas till behandlingen. Enligt vår erfarenhet har administreringen av bikalutamid uppnått signifikant förbättring vid svår akne, men liknande resultat erhölls inte i fall med svår hirsutism. Kosmetiska tillvägagångssätt som laserapplikation och depilatories kan också föreslås för kvinnor som klagar över överdriven eller oönskad hårväxt eller hårbottenförlust (androgen alopeci) (32, 34, 57).
hos patienter med otillräcklig kortisolsekretion efter stimulering, eller om OCPs och antiandrogener inte kan tolereras, rekommenderas en kurs med GCs (57). Data från New et al. ange att oregelbundna menstruationer och akne vänds inom 3 månader efter påbörjandet av glukokortikoidterapin, medan hirsutism kräver nästan 30 månader (59). I motsats till barndomen används ofta långverkande steroider i ungdomar och regimer med 5 mg prednisolon eller 0,25 mg dexametason rekommenderas (53). Men i den verkliga världen kliniska data har visat en mängd olika regimer tillämpas i nccah management (41).
det primära målet med behandlingen bör vara patientens lättnad från de hyperandrogena symtomen. Bland individer med nccah diagnostiserade baserat enbart på laboratorieresultat bör kliniska egenskaper vägleda ledningsrekommendationer, eftersom hormonella och molekylära fynd inte nödvändigtvis förutsäger klinisk svårighetsgrad. Enligt riktlinjerna för det endokrina samhället bör nccah-patienter ges möjlighet att avbryta GC-behandlingen när symtomen försvinner (60). Naturligtvis bör dessa patienter inte gå förlorade för uppföljning, medan behandlingen bör återupptas vid återkommande symtom. Vidare, vid avbrytande, bör patienterna informeras om risken för infertilitet och bör uppmuntras att söka läkare om de vill bli gravida (19). Observera att lämplig överföring av patienten från barn till vuxen endokrinolog bör utföras optimalt efter 1 års synkroniserad övervakning (53, 61).
NCCAH och reproduktion
subfertilitet
subfertilitet rapporteras vanligen i klassiska former av CAH, vilket främst beror på menstruationsstörningar, kronisk anovulation och anatomiska deformiteter. Födelsetalen har uppskattats till 17% jämfört med 65% av kontrollpopulationen (62). Under tiden har data om fertilitet hos kvinnor med NCCAH nyligen utvärderats i detalj och den uppskattade infertilitetsincidensen är 11% bland NCCAH-kvinnor, det vill säga relativt mildare än I CAH, och i många fall är det lätt att lösa. Bidet et al. utvärderad fertilitet hos 190 kvinnor som lider av NCCAH, varav 95 ville bli gravid. I denna befolkning rapporterades 187 graviditeter hos 85 kvinnor, vilket resulterade i 141 födda hos 82 individer. Det måste framhävas att 99 av graviditeterna (52, 9%) inträffade före diagnosen NCCAH, tre av dem med tillämpning av ägglossningsinduktionsprotokoll, resten är spontana, medan 88 graviditeter ägde rum efter nccah-diagnos. I de allra flesta av dem utvecklades graviditeten efter Institutionen för behandling med hydrokortison, medan det hos 11 kvinnor hände spontant (63, 64). I en annan rapport av 22 observerade nccah-kvinnor som önskar graviditet följde 12 graviditeter med prednison (65).
missfall
i kohorten Bidet et al. var missfallsfrekvensen 6, 5 respektive 26, 3% hos patienter som behandlades med GCs respektive obehandlade patienter (64). Resultatet av graviditeten kan till och med vara framgångsrikt utan glukokortikoidbehandling i fall där NCCAH ännu inte diagnostiserades, vilket också rapporterats i en fallrapport från Cuhaci et al. Ändå upplevde samma par två missfall och rapporterade subfertilitet efter denna första graviditet (66). När det gäller möjligheten till prematura graviditeter, som bedömts av Bidet et al., det skiljer sig inte signifikant från resten av befolkningen (15).
Fertilitetsplanering
för de kvinnor med symtomatisk hyperandrogenism eller med rapporterad infertilitet men som vill bli gravida rekommenderas GC-behandling starkt. Under denna period är de använda glukokortikoiderna prednison eller hydrokortison. Den viktigaste åtgärden, eftersom den kommer att styra de efterföljande stegen, är genetisk testning av de blivande föräldrarna. Kvinnor med NCCAH som vill bli gravid bör vara medvetna om risken att föda ett spädbarn med den klassiska formen av sjukdomen. Resultatet av graviditeter bland kvinnor med NCCAH, och mer specifikt förekomsten av spädbarn födda med CCAH, uppskattas i de senaste studierna vara mellan 1, 5 och 2, 5%, med NCCAH på cirka 15% (36, 64). Naturligtvis beror denna frekvens på referenspopulationen, en högre förekomst som förekommer i populationer med höga giftermål (36). De förutspådda chanserna att föräldrar föder ett barn med CAH är 1 av 120 för okänd faderlig genotyp, noll om fadern inte har någon relevant mutation och 1 av 4 om han har en heterozygot mutation (19). Som ett resultat är genetisk testning av dessa kvinnors partners avgörande för att bedöma risken för att föda ett barn med den klassiska formen av CAH (64, 67).
om den blivande mamman bär två NC-mutationer (Tabell 1) finns det inget absolut behov av genetisk testning i den framtida fadern, eftersom han är heterozygot för cyp12a2-mutationerna riskerar avkomman att utveckla den icke-klassiska formen av sjukdomen i framtiden. Det bör dock noteras att bärarstatus är 2-15% i olika populationer och hälften av dessa individer har en allvarlig mutation. Dessutom är definitiva rekommendationer angående situationer där genetisk testning inte krävs svåra med tanke på den ofullkomliga genotyp-fenotypkorrelationen, särskilt för mildare mutationer . Till exempel förekommer p30l-mutationen, oftast associerad med NCCAH, också hos patienter med klassisk CAH . En stor europeisk multicenterstudie visade nyligen att detta var fallet hos 78% av patienterna . I denna studie var den milda v218l-mutationen associerad med klassisk snarare än NCCAH i 30% av fallen. Därför bör alla patienter med NCCAH erbjudas genetisk rådgivning och molekylär bedömning av reproduktiva partners.
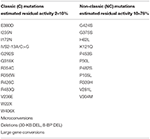
tabell 1. Mutationer av CYP21A2 med minimal (C) eller måttlig (NC) kvarvarande enzymaktivitet (14, 68, 69).
däremot, om den blivande mamman är en sammansatt heterozygot med en svår (C) och en NC-mutation, är genetisk testning av den framtida fadern obligatorisk. Vi bör komma ihåg att data som härrör från studier som använder immunanalyser eller de mer exakta LC-MS/MS-procedurerna konsekvent framhäver oförmågan att på ett tillförlitligt sätt utesluta heterozygositet med basala eller ACTH-stimulerade 17 OHP-värden på grund av den signifikanta överlappningen med det normala intervallet. Man kan föreslå användning av ett Synacthen-test, och om det inte är kompatibelt med heterozygositet (summan av basala och toppstimulerade 17 OHP-värden < 1,5 nmol/L), Kan DNA-testning undvikas. Vi bör dock vara mycket försiktiga vid tolkningen av detta test, eftersom det inte har validerats i andra studier. Därför kan användningen av ACTH stimulerad 21-deoxikortisol antingen enskilt eller som en del av ett steroidförhållande eller steroidprofil underlätta biokemisk identifiering av heterozygoter i framtiden, särskilt som LC-MS/MS blir mer allmänt tillgänglig . Om den blivande Fadern bär en NC-mutation, behövs inget annat. Omvänt, om han är bärare av en klassisk mutation, kommer frågan om prenatal behandling av fostret att uppstå. Alla möjliga genotypkombinationer och de föreslagna procedurerna presenteras i Tabell 2.

tabell 2. Planering av föreslagna förfaranden under graviditeten baserat på blivande föräldrar genotyp.
Prenatal behandling av CAH
syftet med prenatal behandling av CAH är förebyggande av genital virilisering av fostret, men också lindring av stressen som känns av föräldrarna som sannolikt kommer att få ett barn med tvetydiga könsorgan (70). Dexametason används på grund av dess förmåga att korsa placentan och eftersom den inte deaktiveras från placenta 11 ml OH-steroiddehydrogenas och binder endast minimalt till moderns blodkortisolbindande globulin (CBG). Dexametason, genom att undertrycka fetalt ACTH-utsöndring, minskar förhöjd Foster androgenproduktion. Behandlingen måste avbrytas om karyotyp eller DNA-analys avslöjar en man eller en opåverkad kvinna (71). Trots att fostrets genitala virilisering redan har börjat vid 6-7 veckor efter befruktningen, kan korioniska villösa biopsier för genetisk diagnos inte erhållas före den 10: e−12: e veckan. Detta tidsintervall tyder på att alla graviditeter med risk för viriliserande CAH ska behandlas, även om endast 1 av 8 Foster påverkas och kvinnor (19, 34, 70).
huruvida denna exponering av fostret för dexametason för förebyggande åtgärder är medicinskt och etiskt acceptabelt förblir kontroversiellt (70). Djurstudier har visat att dexametason i första trimestern minskar födelsevikt, påverkar betacellens betacellfunktion i njurarna och hjärnans utveckling och predisponerar för ångest, högt blodtryck och hyperglykemi i vuxen ålder (70). Vidare klagar 10-20% av gravida kvinnor som använder dexametasonbehandling under graviditeten på viktökning, ökad aptit, humörsvängningar, sömnlöshet, ödem, högt blodtryck, hyperglykemi och striae. Dock, nya et al. hittade en lägre än genomsnittlig Praderpoäng hos foster som behandlades med dexametason prenatalt, men ingen skillnad i långsiktiga resultat (72). Dessutom, i en recension av Merce Fernandez-Balsells et al., dexametason visade sig vara associerat med minskning av fostervirilisering utan signifikanta maternella eller fetala biverkningar (63). Författarna till ovannämnda granskning samt de på varandra följande riktlinjerna från det endokrina samhället påpekar emellertid att på grund av de små provstorlekarna i hela litteraturens kropp är ämnet fortfarande osäkert och ytterligare utredning behövs tydligt.
beslutet om att inleda behandling bör endast genomföras i stora centra med ett erfaret team och protokoll godkända av institutionella Granskningsnämnder och baserat på familjens värderingar och preferenser och med deras skriftliga informerade samtycke som en förutsättning (19, 63). Flera specialister inom området indikerar att ett högre värde bör läggas på att förhindra onödig prenatal exponering av mor och foster för dexametason snarare än att införa den känslomässiga vägen för tvetydiga könsorgan på föräldrar och patienter. Viktigast av allt bör det klargöras för föräldrar att dexametasonadministration inte ändrar patientstatus utan syftar till att minska behovet av operation snarare än att bevara liv eller intellektuell kapacitet. De måste också veta att sannolikheten för att deras barn kommer att drabbas av den klassiska formen av sjukdomen är hög, trots behandling. Mest avgörande, under tiden, exponering av en ung organism för en mycket potent GC under en särskilt känslig period av fosterprogrammering och tillväxt, vilket mycket väl kan vara värdelöst när det gäller ett foster med XY-karyotypen, stöds för närvarande inte av robusta och obestridliga data.
helst bör det finnas en tidig diagnos, detta utförs via en icke-invasiv teknik och före början av könsviriliseringen av fostret. Arbeta i denna riktning pekar nya studier på användningen av cellfritt foster-DNA erhållet från moderplasma som en lovande metod som möjliggör bestämning av fosterkön och diagnos av CAH vid en tidig graviditetsålder (<9 veckor). Om denna procedur genomförs i stor utsträckning i klinisk praxis, kommer onödig prenatal dexametasonbehandling att undvikas (73).
behandling under graviditet
under graviditet bör kvinnor med NCCAH helst behandlas med hydrokortison, inte bara för att graviditet vanligtvis åtföljs av förhöjda stressnivåer, men också som en förebyggande åtgärd mot ovan nämnda höga förekomst av missfall. Denna form av glukokortikoid är gynnad på grund av dess metabolisering av placental enzym 11 ml OH steroiddehydrogenas II, vilket förhindrar glukokortikoid från att ha någon effekt på fostret (71). Vid tidpunkten för uppfattningen bör hydrokortisondosen ökas till 20-25 mg/dag och dosändringar utförs var 6-8 veckor i syfte att hålla testosteronnivåerna vid de övre normala nivåerna av graviditetstrimestern (74). Som ett exempel visas testosteron-och D4-värdena hos en patient med NCCAH från uppfattningen upp till 3 år efter leverans av två friska tvillingar (Figur 1).
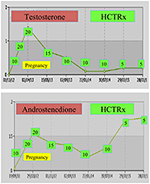
Figur 1. Testosteron-och andostenedionnivåer och efterföljande hydrokortisondosering (HCTRx), från befruktning till 3 år efter leverans administrerad till en patient med NCCAH.
andra speciella problem
stresshantering i NCCAH
under större livshotande stress, kirurgi eller allvarlig sjukdom kan patienter med nccah som behandlas med glukokortikoid kräva större eller mer frekventa doser glukokortikoider med tanke på att deras binjurefunktion är iatrogeniskt undertryckt. Det är därför viktigt att utbilda föräldrar till små barn, samt att utbilda patienter om deras övergång till vuxenvård, om stressdosering. För nccah-patienter som inte behandlas med GCs eller i händelse av avbrytande, bör kortisolsvar på ACTH bedömas. Nästan en tredjedel av nccah-patienterna svarar otillräckligt på stimuleringen (15, 75). För dem som svarar normalt på ACTH-stimulering rekommenderas ingen behandling med stressdoser (19). Mineralokortikoidbehandling krävs inte i något av fallen, eftersom dessa patienter har normal aldosteronsekretion och inte utvecklar saltavfall (58). Observera att vid hypertyreoidism eller en ökning av levotyroxinbehandling ökar kortisolclearance och en binjurskris kan uppstå (58, 76).
psykobiologiska aspekter i NCCAH
Meyer-Bahlburg et al. har i flera studier rapporterat nedsatt psykologisk profil bland patienter med NCCAH på grund av 21oh-brist. Mer specifikt hade de drabbade kvinnorna en högre maskulinisering / defeminiseringspoäng på flera mått på könsrelaterat beteende jämfört med normala kontrollkvinnor, men markant mindre än hos kvinnor med klassisk CAH. De hade också signifikant ökad bisexuell eller homosexuell orientering. Retrospektiva kliniska kvalitativa intervjuer med dessa kvinnor avslöjade en historia av obehag och social stress relaterad till deras förbehandlingsupplevelser med androgenberoende tecken, såsom akne, hirsutism och uppfattningssvårigheter (59, 77, 78). Liknande, Arlt et al. visade att subjektiv hälsotillstånd försämrades signifikant över alla åtta domäner i en kortsiktig hälsoundersökning, med de mest framträdande skillnaderna, jämfört med ålders – och könsmatchade kontroller, relaterade till domänerna allmän hälsa, vitalitet och rollbegränsningar på grund av känslomässiga problem. Frågeformuläret för Sjukhusangst och Depression (HADS) avslöjade också ökade ångestpoäng (41). Med tanke på dessa resultat bör psykologiska parametrar för att vägleda terapi övervägas hos kvinnor med NCCAH och i samband med patientorienterat tillvägagångssätt måste en psykologisk diagnos och stöd erbjudas.
slutsatser
NCCAH anses vara den mer frekventa och mildare formen av CAH på grund av retention av 20-50% enzymaktivitet. De flesta patienter kan söka medicinsk rådgivning i något skede av sitt liv på grund av symtom relaterade till androgenöverskott. Dessa inkluderar för tidig pubarche, snabb tillväxt, hirsutism, akne, menstruella oegentligheter eller infertilitet, liksom många andra mindre framträdande manifestationer. Tidiga morgonvärden på 17 OHP som ett bra initialt screeningtest och ytterligare utvärdering med ACTH-stimulering och, i fallet med gränsresultat, genetisk testning, rekommenderas. Som en allmän regel bör androstenedion och inte 17 OHP-nivåer användas under uppföljningen.
behandlingsbeslutet bör baseras på bedömning av fakta och bör följa ett individualiserat tillvägagångssätt. Behandling är inte alltid indicerad om inte patienten är symptomatisk, till exempel barn med tidig debut och snabb progression av skönhets-och kroppshår, snabb tillväxt och/eller skelettutveckling eller kvinnor med oligomenorrhea, akne, hirsutism, infertilitet eller en kombination av dessa och andra av ovan nämnda symtom. Genetisk rådgivning rekommenderas starkt hos nccah-kvinnor som vill bli gravida, liksom genotypning av fadern. Den övergripande hanteringen av patienten innefattar dessutom hantering av de sannolika komplikationerna av glukokortikoidterapi eller metabolismrelaterade manifestationer av sjukdomen. Slutligen, med tanke på att många terapeutiska frågor relaterade till lämplig hantering av dessa patienter ännu inte har klarlagts, är det mycket viktigt för den behandlande läkaren att hålla sig uppdaterad med all utveckling på detta område och att integrera de nya uppgifterna i hans kliniska praxis. Visst är ytterligare kliniska studier på detta område väsentliga.
för att sammanfatta, för nccah-kvinnan, är det perfekta tillvägagångssättet skräddarsytt, som innehåller en smidig övergång av hennes ledning när hon hänvisas från barn till vuxen endokrinolog, tillsammans med symptomorienterad behandling som kommer att följa med henne under hela sitt liv. Med tanke på de många faktorer som påverkar det hyperandrogena systemet är det lämpligt att uppmuntra patienter att anta en hälsosammare livsstil genom att förbättra sina kostvanor, öka träningen och sikta på viktminskning. Dessutom är psykologiskt stöd i vissa fall ofta fördelaktigt. Dessutom behöver specialister inom områden som är involverade i behandlingen av denna sjukdom, såsom hudläkare, gynekologer och psykologer, fördjupad förståelse för hanteringen av misstänkta eller redan diagnostiserade fall av NCCAH. Avslutningsvis, låt oss komma ihåg det insiktsfulla citatet från den amerikanska astronomen Vera Cooper Rubin: ”vetenskapen utvecklas bäst när observationer tvingar oss att ändra våra förutfattade meningar.”
Författarbidrag
alla listade författare har gjort ett väsentligt, direkt och intellektuellt bidrag till arbetet och godkänt det för publicering.
intressekonflikt uttalande
författarna förklarar att forskningen genomfördes i avsaknad av kommersiella eller finansiella relationer som kan tolkas som en potentiell intressekonflikt.
1. Speiser PW, Vit PC. Medfödd adrenal hyperplasi. N Engl J Med. (2003) 349:776–88. doi: 10.1056 / NEJMra021561
PubMed Abstrakt / CrossRef fulltext / Google Scholar
2. Turcu AF, Auchus RJ. Adrenal steroidogenesis and congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. (2015) 44:275–96. doi: 10.1016/j.ecl.2015.02.002
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Speiser PW. Nonclassic adrenal hyperplasia. Rev Endocr Metab Disord. (2009) 10:77–82. doi: 10.1007/s11154-008-9097-x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Det finns många olika typer av produkter. Hundra års kongenital adrenal hyperplasi i Sverige: en retrospektiv, populationsbaserad kohortstudie. Lancet Diabetes Endocrinol. (2013) 1:35–42. doi: 10.1016/S2213-8587(13)70007-x
PubMed Abstrakt | CrossRef fulltext | Google Scholar
5. Dupont B, Rubinstein P, Piazza A, Kastelan A, Nya MI. Hög frekvens av icke-klassisk steroid 21-hydroxylasbrist. Är J Hum Genet. (1985) 37:650–67.
PubMed Abstract | Google Scholar
6. Wilson RC, Nimkarn S, Dumic M, Obeid J, Azar MR, Azar M, et al. Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Mol Genet Metab. (2007) 90:414–21. doi: 10.1016/j.ymgme.2006.12.005
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Miller WL. Clinical review 54: genetics, diagnosis, and management of 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1994) 78:241–6. doi: 10.1210/jcem.78.2.8106606
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Witchel SF, Azziz R. Nonclassic congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2010) 2010:625105. doi: 10.1186/1687-9856-2010-625105
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Wilson RC, Mercado AB, Cheng KC, nya MI. Steroid 21-hydroxylasbrist: genotyp kan inte förutsäga fenotyp. J Clin Endocrinol Metab. (1995) 80:2322–9. doi: 10.1210 / jc.80.8.2322
CrossRef fulltext/Google Scholar
10. Vit PC. Neonatal screening för medfödd adrenal hyperplasi. Nat Rev Endocrinol. (2009) 5:490–8. doi: 10.1038/nrendo.2009.148
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Kohn B, Levine LS, Pollack MS, Pang S, Lorenzen F, Levy D, et al. Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. (1982) 55:817–27. doi: 10.1210/jcem-55-5-817
PubMed Abstract | CrossRef Full Text | Google Scholar
12. C, Azziz R, Carmina E, Dewailly D, Fruzzetti F, iba Biseez L, et al. 21-Hydroxylasbrist icke-klassisk adrenal hyperplasi är en progressiv sjukdom: en multicenterstudie. Är J Förlossningsläkare Gynecol. (2000) 183:1468–74. doi: 10.1067 / mob.2000.108020
PubMed Abstrakt / CrossRef fulltext / Google Scholar
13. Azziz R, Hincapie LA, Knochenhauer ES, Dewailly D, räv L, Stövlar LR. Screening för 21-hydroxylasbrist icke-klassisk adrenal hyperplasi bland hyperandrogena kvinnor: En prospektiv studie. Fertil Steril. (1999) 72:915–25. doi: 10.1016/S0015-0282(99)00383-0
PubMed Abstrakt | CrossRef fulltext | Google Scholar
14. Det finns också ett stort antal av dem. Spektrumet av kliniska, hormonella och molekylära fynd hos 280 individer med icke-klassisk medfödd adrenal hyperplasi orsakad av mutationer av cyp21a2-genen. Clin Endocrinol (Oxf). (2015) 82:543–9. doi: 10.1111 / cen.12543
PubMed Abstrakt / CrossRef fulltext / Google Scholar
15. Bidet m, Bellann C-Chantelot C, Galand-Portier M-B, Tardy V, Billaud L, Laborde K, et al. Klinisk och molekylär karakterisering av en kohort av 161 orelaterade kvinnor med icke-klassisk medfödd adrenal hyperplasi på grund av 21-hydroxylasbrist och 330 familjemedlemmar. J Clin Endocrinol Metab. (2009) 94:1570–8. doi: 10.1210 / jc.2008-1582
PubMed Abstrakt / CrossRef fulltext / Google Scholar
16. Escobar-Morreale HF, Sanch Ubign R, San Mill Ubign JL. En prospektiv studie av förekomsten av icke-klassisk medfödd adrenal hyperplasi bland kvinnor som uppvisar hyperandrogena symtom och tecken. J Clin Endocrinol Metab. (2008) 93:527–33. doi: 10.1210 / jc.2007-2053
PubMed Abstrakt / CrossRef fulltext / Google Scholar
17. Dewailly D, Vantyghem-HAUDIQUET MC, Sainsard C, Buvat J, Cappoen JP, Ardaens K, et al. Kliniska och biologiska fenotyper vid sen debut 21-hydroxylasbrist. J Clin Endocrinol Metab. (1986) 63:418–23. doi: 10.1210 / jcem-63-2-418
PubMed Abstrakt / CrossRef fulltext / Google Scholar
18. G ubicn ubic EN, Oz Ubicn ZA, Alika Ubiifoglu A, Engiz O, Bulum B, Kandemir N. (2011). Är basal serum 17-OH progesteron en pålitlig parameter för att förutsäga icke-klassisk medfödd adrenal hyperplasi i för tidig adrenarche? Turk J Pediatric. 53:274–80.
PubMed Abstract / Google Scholar
19. Det är en av de mest populära och mest populära. Medfödd adrenal hyperplasi på grund av steroid 21-hydroxylasbrist: en endokrin samhälle klinisk praxis riktlinje. J Clin Endocrinol Metab. (2010) 95:4133–60. doi: 10.1210 / jc.2009-2631
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Dacou-Voutetakis C, Dracopoulou M. High incidence of molecular defects of the CYP21 gene in patients with premature adrenarche. J Clin Endocrinol Metab. (1999) 84:1570–4. doi: 10.1210/jc.84.5.1570
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Jag är en av de mest kända. Diagnosen av icke-klassisk medfödd hyperplasi i binjurebarken på grund av 21-hydroxylasbrist, baserad på serumbasal eller post-ACTH stimulering 17-hydroxiprogesteron, kan leda till falsk positiv diagnos. Clin Endocrinol (Oxf). (2016) 84:23–9. doi: 10.1111 / pris 12935
PubMed Abstrakt / CrossRef fulltext / Google Scholar
22. Jag är en av de mest kända och mest kända. LC-MS / MS förbättrar screening mot 21-hydroxylasbrist. Gynecol Endocrinol. (2015) 31:296–300. doi: 10.3109/09513590.2014.994599
PubMed Abstrakt | CrossRef fulltext | Google Scholar
23. bbc, CC, Goldman MM, Zhang K, Clark-N, Reitz RE Welt CK. Samtidig mätning av tretton steroidhormoner hos kvinnor med polycystiskt äggstockssyndrom och kontrollkvinnor som använder vätskekromatografi-tandemmasspektrometri. PLoS Ett. (2014) 9:e93805. doi: 10.1371 / tidskrift.pone.0093805
PubMed Abstrakt / CrossRef fulltext / Google Scholar
24. Ray JA, Kushnir MM, Yost RA, Rockwood AL, Wayne Meikle A. prestandaförbättring vid mätning av 5 endogena steroider med LC-MS / MS kombinerat med differentiell jonmobilitetsspektrometri. Clin Chim Acta. (2015) 438:330–6. doi: 10.1016 / j. cca.2014.07.036
PubMed Abstrakt / CrossRef fulltext / Google Scholar
25. Det finns många olika typer av produkter. Otillräckligt kortisolrespons på tetrakosaktid (Synacthen-testet) i icke-klassisk medfödd adrenal hyperplasi: ett undantag från regeln? Horm Res Paediatr. (2015) 83:262–7. doi: 10.1159/000369901
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. (1992) 90:584–95. doi: 10.1172/JCI115897
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Temeck JW, Pang SY, Nelson C, nya MI. Genetiska defekter av steroidogenes i för tidig pubarche. J Clin Endocrinol Metab. (1987) 64:609–17. doi: 10.1210 / jcem-64-3-609
PubMed Abstrakt / CrossRef fulltext / Google Scholar
28. Voutilainen r, J. för tidig Adrenarche: etiologi, kliniska fynd och konsekvenser. J Steroid Biochem Mol Biol. (2015) 145:226–36. doi: 10.1016/j.jsbmb.2014.06.004
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Oberfield SE, Sopher AB, Gerken AT. Approach to the girl with early onset of pubic hair. J Clin Endocrinol Metab. (2011) 96:1610–22. doi: 10.1210/jc.2011-0225
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Janus D, Wojcik M, Malunowicz E, Starzyk JB. Ett fall av återkommande labiala vidhäftningar hos ett 15 månader gammalt barn med asymptomatisk icke-klassisk medfödd adrenal hyperplasi på grund av 21-hydroxylasbrist. J Pediatr Endocrinol Metab. (2012) 25:1017–21. doi: 10.1515 / jpem-2012-0190
PubMed Abstrakt | CrossRef fulltext/Google Scholar
31. Gopal-Kothandapani JS, Petkar A, O ’ Shea E, Banerjee I. perianalt hår som en ovanlig presentation av icke-klassisk medfödd adrenal hyperplasi. BMJ Case rep. (2013) 2013:bcr2013010123. doi: 10.1136/bcr-2013-010123
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Falhammar H, Nordenström A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine. (2015) 50:32–50. doi: 10.1007/s12020-015-0656-0
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Chrousos GP, Loriaux DL, Mann DL, Cutler GB. Late-onset 21-hydroxylase deficiency mimicking idiopathic hirsutism or polycystic ovarian disease. Ann Intern Med. (1982) 96:143–8. doi: 10.7326/0003-4819-96-2-143
CrossRef Full Text | Google Scholar
34. Witchel SF. Non-classic congenital adrenal hyperplasia. Steroids. (2013) 78:747–50. doi: 10.1016/j.steroids.2013.04.010
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Carmina E, Rosato F, Jannì A, Rizzo M, Longo RA. Extensive clinical experience: relative prevalence of different androgen excess disorders in 950 women referred because of clinical hyperandrogenism. J Clin Endocrinol Metab. (2006) 91:2–6. doi: 10.1210/jc.2005-1457
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Det är en av de mest kända. Reproduktivt resultat av kvinnor med 21-hydroxylasbrist icke-klassisk adrenal hyperplasi. J Clin Endocrinol Metab. (2006) 91:3451–6. doi: 10.1210 / jc.2006-0062
PubMed Abstrakt / CrossRef fulltext / Google Scholar
37. Levin JH, Carmina E, Lobo RA. Är den olämpliga gonadotropinsekretionen hos patienter med polycystiskt äggstockssyndrom liknande den hos patienter med medfödd adrenal hyperplasi hos vuxna? Fertil Steril. (1991) 56:635–40. doi: 10.1016/S0015-0282(16)54592-0
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Carmina E, Lobo RA. Ovarian suppression reduces clinical and endocrine expression of late-onset congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Fertil Steril. (1994) 62:738–43. doi: 10.1016/S0015-0282(16)56998-2
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Ludwig E. klassificering av de typer av androgenetisk alopeci (vanlig skallighet) som förekommer hos honkön. Br J Dermatol. (1977) 97:247–54. doi: 10.1111 / j. 1365-2133. 1977.tb15179.x
PubMed Abstrakt / CrossRef fulltext / Google Scholar
40. Trakakis E, Papadavid E, Dalamaga M, Koumaki D, Stavrianeas N, Rigopoulos D, et al. Prevalens av icke-klassisk medfödd adrenal hyperplasi på grund av 21-hydroxylasbrist hos grekiska kvinnor med akne: en sjukhusbaserad tvärsnittsstudie. J Eur Acad Dermatol Venereol. (2013) 27:1448–51. doi: 10.1111 / j. 1468-3083. 2012.04613.x
PubMed Abstrakt / CrossRef fulltext / Google Scholar
41. Arlt W, Willis DS, Wild SH, Krone N, Doherty EJ, Hahner S, et al. Hälsotillstånd hos vuxna med medfödd adrenal hyperplasi: en kohortstudie av 203 patienter. J Clin Endocrinol Metab. (2010) 95:5110–21. doi: 10.1210 / jc.2010-0917
PubMed Abstrakt / CrossRef fulltext / Google Scholar
42. Saygili F, Oge A, Yilmaz C. hyperinsulinemi och insulinkänslighet hos kvinnor med icke-klassisk medfödd adrenal hyperplasi på grund av 21-hydroxylasbrist: förhållandet mellan serumleptinnivåer och kronisk hyperinsulinemi. Horm Res. (2005) 63:270-4. doi: 10.1159/000086363
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Speiser PW, Serrat J, New MI, Gertner JM. Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1992) 75:1421–4. doi: 10.1210/jc.75.6.1421
PubMed Abstract | CrossRef Full Text | Google Scholar
44. Han TS, Stimson RH, Rees DA, Krone N, Willis DS, Conway GS, et al. Glukokortikoid behandlingsregim och hälsoresultat hos vuxna med medfödd adrenal hyperplasi. Clin Endocrinol. (2013) 78:197–203. doi: 10.1111 / cen.12045
PubMed Abstrakt / CrossRef fulltext / Google Scholar
45. Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Användning av orala kortikosteroider och risk för frakturer. J Ben Miner Res. (2000) 15:993-1000. doi: 10.1359/jbmr.2000.15.6.993
PubMed Abstract | CrossRef Full Text | Google Scholar
46. Nebesio TD, Renbarger JL, Nabhan ZM, Ross SE, Slaven JE, Li L, et al. Differential effects of hydrocortisone, prednisone, and dexamethasone on hormonal and pharmacokinetic profiles: a pilot study in children with congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2016) 2016:17. doi: 10.1186 / s13633-016-0035-5
PubMed Abstrakt / CrossRef fulltext / Google Scholar
47. Weintrob N, Dickerman Z, Sprecher E, Galatzer a, Pertzelan A. icke-klassisk 21-hydroxylasbrist i spädbarn och barndom: effekten av behandlingstiden på puberteten och sluthöjden. Eur J Endocrinol. (1997) 136:188–95. doi: 10.1530/eje.0.1360188
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Bonfig W, Bechtold S, Schmidt H, Knorr D, Schwarz HP. Reduced final height outcome in congenital adrenal hyperplasia under prednisone treatment: deceleration of growth velocity during puberty. J Clin Endocrinol Metab. (2007) 92:1635–9. doi: 10.1210/jc.2006-2109
PubMed Abstract | CrossRef Full Text | Google Scholar
49. Dörr HG. Tillväxt hos patienter med klassisk medfödd adrenal hyperplasi på grund av 21-hydroxylasbrist. Horm Res. (2007) 68(Suppl 5): 93-9. doi: 10.1159 / 000110587
PubMed Abstrakt / CrossRef fulltext / Google Scholar
50. Hoepffner W, Kaufhold a, Willgerodt H, Keller E. patienter med klassisk medfödd adrenal hyperplasi på grund av 21-hydroxylasbrist kan uppnå sin målhöjd: Leipzig-upplevelsen. Horm Res. (2008) 70:42-50. doi: 10.1159/000129677
PubMed Abstract | CrossRef Full Text | Google Scholar
51. van der Kaay D, van den Akker E. Ultralow-dose dexamethasone to preserve endogenous cortisol stress response in nonclassical congenital adrenal hyperplasia: a new promising treatment. Int J Endocrinol Metab. (2014) 12:e14657. doi: 10.5812/ijem.14657
CrossRef Full Text | Google Scholar
52. Lin-Su K, Vogiatzi MG, Marshall I, Harbison MD, Macapagal MC, Betensky B, et al. Treatment with growth hormone and luteinizing hormone releasing hormone analog improves final adult height in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. (2005) 90:3318–25. doi: 10.1210/jc.2004-2128
PubMed Abstract | CrossRef Full Text | Google Scholar
53. McCann-Crosby B, Chen M-J, Lyons SK, Lin Y, Axelrad M, Dietrich JE, et al. Nonclassical congenital adrenal hyperplasia: mål för behandling och övergång. Pediatric Endocrinol Rev. (2014) 12:224-38.
PubMed Abstract / Google Scholar
54. Det är en av de mest populära och mest populära. Medfödd adrenal hyperplasi hos vuxna: en översyn av medicinska, kirurgiska och psykologiska problem. Clin Endocrinol. (2006) 64:2–11. doi: 10.1111 / j. 1365-2265.2005. 02410.x
PubMed Abstract | CrossRef Full Text | Google Scholar
55. Mah PM, Jenkins RC, Rostami-Hodjegan A, Newell-Price J, Doane A, Ibbotson V, et al. Weight-related dosing, timing and monitoring hydrocortisone replacement therapy in patients with adrenal insufficiency. Clin Endocrinol. (2004) 61:367–75. doi: 10.1111/j.1365-2265.2004.02106.x
PubMed Abstract | CrossRef Full Text | Google Scholar
56. Diaz A, Laufer Mr Breech LLAA av PC på A, Vård AC av O och GC på AH. Menstruation hos flickor och ungdomar: användning av menstruationscykeln som ett viktigt tecken. Pediatrik. (2006) 118:2245–50. doi: 10.1542 / peds.2006-2481
CrossRef fulltext | Google Scholar
57. Merke DP, Poppas DP. Hantering av ungdomar med medfödd adrenal hyperplasi. lancet Diabetes Endocrinol. (2013) 1:341–52. doi: 10.1016/S2213-8587(13)70138-4
PubMed Abstract | CrossRef Full Text | Google Scholar
58. Verma S, Green-Golan L, VanRyzin C, Drinkard B, Mehta SP, Weise M, et al. Adrenomedullary function in patients with nonclassic congenital adrenal hyperplasia. Horm Metab Res. (2010) 42:607–12. doi: 10.1055/s-0030-1253385
PubMed Abstract | CrossRef Full Text | Google Scholar
59. New MI. Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2006) 91:4205–14. doi: 10.1210/jc.2006-1645
PubMed Abstract | CrossRef Full Text | Google Scholar
60. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2018) 103:4043–88. doi: 10.1210/jc.2018-01865
PubMed Abstract | CrossRef Full Text | Google Scholar
61. Kruse B, Riepe FG, Krone N, Bosinski HAG, Kloehn S, Partsch CJ, et al. Congenital adrenal hyperplasia – how to improve the transition from adolescence to adult life. Exp Clin Endocrinol Diabetes. (2004) 112:343–55. doi: 10.1055/s-2004-821013
PubMed Abstract | CrossRef Full Text | Google Scholar
62. De flesta av oss har inte haft några problem. Nedsatt sexuella och reproduktiva resultat hos kvinnor med klassiska former av medfödd adrenal hyperplasi. J Clin Endocrinol Metab. (2007) 92:1391–6. doi: 10.1210 / jc.2006-1757
PubMed Abstrakt / CrossRef fulltext / Google Scholar
63. Det finns många olika typer av produkter. Prenatal dexametason användning för förebyggande av virilisering vid graviditeter med risk för klassisk medfödd adrenal hyperplasi på grund av 21-hydroxylas (CYP21A2) brist: en systematisk granskning och metaanalyser. Clin Endocrinol. (2010) 73:436–44. doi: 10.1111 / j. 1365-2265.2010. 03826.x
CrossRef fulltext/Google Scholar
64. Bidet m, Bellann C-Chantelot C, Galand-Portier MB, Golmard JL, Tardy V, Morel Y, et al. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2010) 95:1182–90. doi: 10.1210/jc.2009-1383
PubMed Abstract | CrossRef Full Text | Google Scholar
65. Birnbaum MD, Rose LI. Late onset adrenocortical hydroxylase deficiencies associated with menstrual dysfunction. Obstet Gynecol. (1984) 63:445–51.
PubMed Abstract | Google Scholar
66. Cuhaci N, Aydin C, Yesilyurt A, Pinarli FA, Ersoy R, Cakir B. Nonclassical congenital adrenal hyperplasia and pregnancy. Case Rep Endocrinol. (2015) 2015:296924. doi: 10.1155/2015/296924
PubMed Abstract | CrossRef Full Text | Google Scholar
67. Deneux C, Tardy V, Anne D, Mornet E, Billaud L, Charron D, et al. Fenotyp-genotyp korrelation hos 56 kvinnor med icke-klassisk medfödd adrenal hyperplasi på grund av 21-hydroxylasbrist. J Clin Endocrinol Metab. (2001) 86:207–13. doi: 10.1210/jcem.86.1.7131
PubMed Abstrakt / CrossRef fulltext / Google Scholar
68. Simonetti l, Bruque CD, Ormbunke C, Benavides-Mori B, Delea M, Kolomenski JE, et al. Cyp21a2 mutation update: omfattande analys av databaser och publicerade genetiska varianter. Hum Mutat. (2018) 39:5–22. doi: 10.1002 / humu.23351
PubMed Abstract | CrossRef Full Text | Google Scholar
69. OMIM Entry – * 613815 – CYTOCHROME P450 FAMILY 21 SUBFAMILY A POLYPEPTIDE 2; CYP21A2.
Google Scholar
70. Miller WL, Witchel SF. Prenatal treatment of congenital adrenal hyperplasia: risks outweigh benefits. Am J Obstet Gynecol. (2013) 208:354–9. doi: 10.1016/j.ajog.2012.10.885
PubMed Abstract | CrossRef Full Text | Google Scholar
71. Lekarev O, New MI. Adrenal disease in pregnancy. Best Pract Res Clin Endocrinol Metab. (2011) 25:959–73. doi: 10.1016/j.beem.2011.08.004
PubMed Abstract | CrossRef Full Text | Google Scholar
72. New MI, Abraham M, Yuen T, Lekarev O. An update on prenatal diagnosis and treatment of congenital adrenal hyperplasia. Semin Reprod Med. (2012) 30:396–9. doi: 10.1055/s-0032-1324723
PubMed Abstract | CrossRef Full Text | Google Scholar
73. Kazmi D, Bailey J, Yau M, Abu-Amer W, Kumar A, Low M, et al. New developments in prenatal diagnosis of congenital adrenal hyperplasia. J Steroid Biochem Mol Biol. (2017) 165:121–3. doi: 10.1016/j.jsbmb.2016.06.016
PubMed Abstract | CrossRef Full Text | Google Scholar
74. Det är en av de mest populära och mest populära. Konsensus uttalande om 21-hydroxylasbrist från European Society for Pediatric Endocrinology och lawson wilkins pediatric endocrine society. Horm Res. (2002) 58:188-95. doi: 10.1159 / 000065490
PubMed Abstrakt | CrossRef fulltext/Google Scholar
75. Nandagopal R, Sinaii N, Avila NA, Van Ryzin C, Chen W, Finkielstain GP, et al. Fenotypisk profilering av föräldrar med kryptisk icke-klassisk medfödd adrenal hyperplasi: fynd i 145 orelaterade familjer. Eur J Endocrinol. (2011) 164:977–84. doi: 10.1530 / EJE-11-0019
PubMed Abstrakt | CrossRef fulltext/Google Scholar
76. Takasu N, Nakachi K, Higa H. utveckling av Graves hypertyreoidism orsakade en binjurskris hos en patient med tidigare okänd icke-klassisk 21-hydroxylasbrist. Praktikant Med. (2010) 49:1395–400. doi: 10.2169 / internmedicin.49.3573
PubMed Abstract | CrossRef Full Text | Google Scholar
77. Meyer-Bahlburg HFL, Dolezal C, Baker SW, Carlson AD, Obeid JS, New MI. Prenatal androgenization affects gender-related behavior but not gender identity in 5-12-year-old girls with congenital adrenal hyperplasia. Arch Sex Behav. (2004) 33:97–104. doi: 10.1023/B:ASEB.0000014324.25718.51
CrossRef Full Text | Google Scholar
78. Meyer-Bahlburg HFL, Dolezal C, Baker SW, nya MI. Sexuell läggning hos kvinnor med klassisk eller icke-klassisk medfödd adrenal hyperplasi som en funktion av graden av prenatalt androgenöverskott. Arch Sex Beter Sig. (2008) 37:85–99. doi: 10.1007 / s10508-007-9265-1
PubMed Abstrakt / CrossRef fulltext / Google Scholar