La sclérotique bleue est causée par la minceur et la transparence des fibres de collagène de la sclérotique qui permettent la visualisation de l’uvée sous-jacente (Figure 1). La sclérotique est la couche externe blanche de l’œil, entourant l’iris peut être amincie dans les maladies congénitales telles que l’ostéogenèse imparfaite 1). D’autres maladies associées à la sclérotique bleue comprennent de multiples troubles du tissu conjonctif, tels que le syndrome de la cornée fragile 2), le syndrome de Marshall et de Stickler 3), le syndrome de POEMS (polyneuropathie, organomégalie, endocrinopathie, protéines monoclonales et modifications cutanées) 4), le syndrome de Marfan, le syndrome d’Ehlers–Danlos, le pseudoxanthome elasticum et le syndrome de Willems De Vries, pour n’en nommer que quelques-uns. Les troubles osseux et sanguins figurent également sur la liste, notamment l’anémie de Diamond–Blackfan, l’anémie ferriprive sévère, la maladie de Paget juvénile et le déficit en phosphatase acide 5).
Les formes sévères d’ostéogenèse imparfaite sont le plus souvent diagnostiquées tôt dans la vie, mais les cas bénins peuvent ne pas être notés avant plus tard dans la vie. La couleur bleu-gris de la sclérotique est due aux veines choroïdiennes sous-jacentes qui se manifestent à travers. Cela est dû au fait que la sclérotique est plus mince que la normale car le collagène de type I défectueux ne se forme pas correctement 6). La sclérotique est une structure de tissu conjonctif dense et mal vascularisée composée de collagène de types I, III, IV, V, VI et VIII, ainsi que d’élastine, de protéoglycanes et de glycoprotéines 7).
Aux États-Unis, l’incidence de l’ostéogenèse imparfaite est estimée à une pour 20 000 naissances vivantes. On estime que 20 000 à 50 000 personnes sont touchées par l’ostéogenèse imparfaite aux États-Unis 8).
Les personnes atteintes d’ostéogenèse imparfaite naissent avec un tissu conjonctif défectueux ou sans capacité de le fabriquer, généralement en raison d’une carence en collagène de type I. Cette carence provient d’une substitution d’acides aminés de la glycine par des acides aminés plus volumineux dans la structure en triple hélice du collagène. En conséquence, le corps peut réagir en hydrolysant la structure de collagène incorrecte 9). Si le corps ne détruit pas le collagène inapproprié, la relation entre les fibrilles de collagène et les cristaux d’hydroxyapatite pour former l’os est altérée, provoquant une fragilité. En tant que trouble génétique, l’ostéogenèse imparfaite a toujours été considérée comme un trouble autosomique dominant du collagène de type I. La plupart des cas ont été causés par des mutations dans les gènes COL1A1 et COL1A2 10). Au cours des dernières années, il y a eu l’identification de formes autosomiques récessives. Au moins sept sous-ensembles ont été décrits, bien que quatre sous-types principaux soient les plus courants et varient de légers à sévères. Les individus atteints d’ostéogenèse imparfaite de type I ont une faible déformation osseuse, une sclérotique bleue persistante, une taille proche de la normale à l’âge adulte et une > 50% de chance de perte auditive à l’âge adulte. Les patients présentant une ostéogenèse imparfaite périnatale létale (type II) présentent la plus grande gravité, avec de multiples fractures in utero ou à partir de l’accouchement. Ces patients sont généralement mort-nés ou meurent tôt. La gravité de l’ostéogenèse imparfaite dépend du défaut génétique spécifique. La plupart des cas d’ostéogenèse imparfaite sont hérités d’un parent. Cependant, certains cas sont le résultat de nouvelles mutations génétiques. Une personne atteinte d’ostéogenèse imparfaite a 50% de chances de transmettre le gène et la maladie à ses enfants 11).
Figure 1. Anatomie oculaire
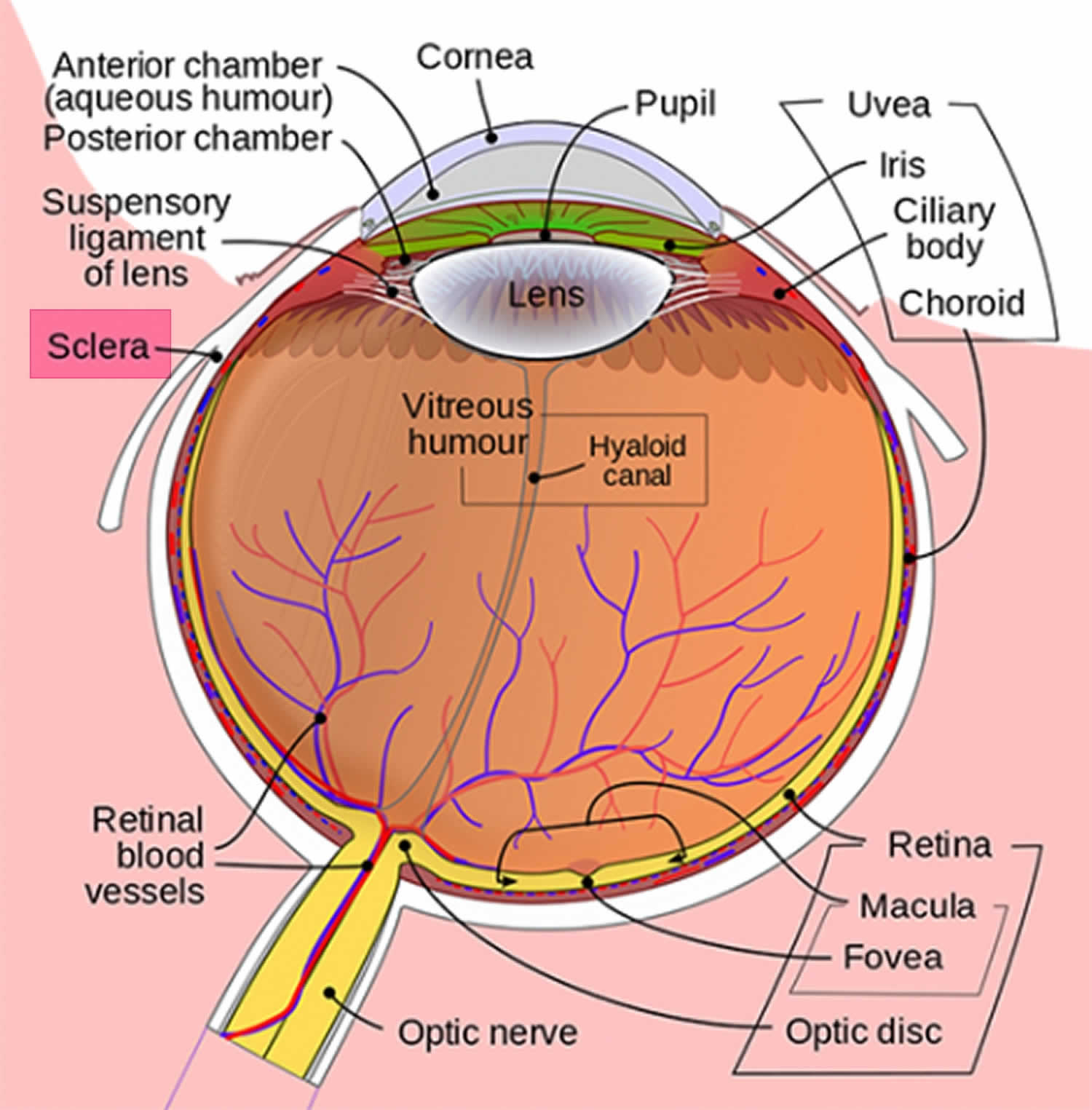
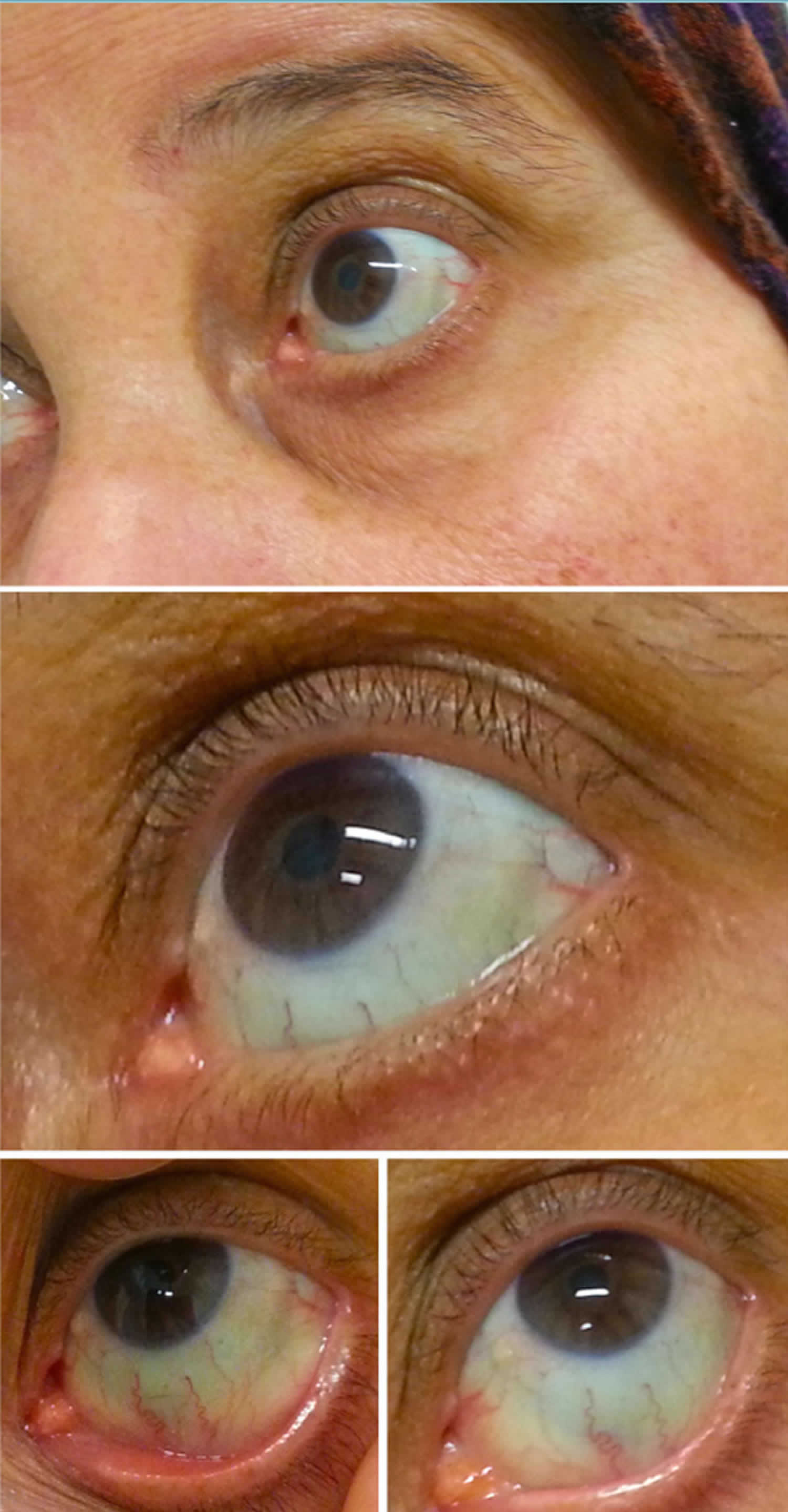
Figure 2. Sclérotique bleue
Causes de la sclérotique bleue
La sclérotique bleue est la manifestation la plus cohérente de l’ostéogenèse imparfaite, qui résulte d’une mutation dans COLIA1 et COL1A2, codant pour le procollagène de type I. Cependant, les classifications de cette affection (types IV-VI) ont été identifiées avec des scléroses normales. L’ostéogenèse imparfaite est également associée à une fragilité anormale des os et à une surdité.
La cornée cassante, la sclérotique bleue et les cheveux roux sont associés au syndrome de la cornée cassante, une affection qui présente également des anomalies squelettiques, dentaires et cutanées 13). Une mutation faux sens dans le ZNF469 s’est avérée être responsable de la maladie.
D’autres conditions sous-jacentes associées à la formation de la sclérotique bleue comprennent le syndrome d’Ehlers-Danlos (type VI), le pseudoxanthome élastique, la cornée plane, la sclérocorne périphérique, la buphtalmie, le kératocône, le kératoglobus, une myopie élevée, un staphylome ciliaire / équatorial, une mélanocytose oculodermique et une microcornée. Rarement, la sclérotique bleue survient avec le syndrome de Hallermann-Streiff, le syndrome de Marfan, le syndrome de Turner, le syndrome de Cheney, le syndrome de Menkes, la pyknodysostose, les cornées cassantes ou la dysplasie ectodermique.
Une sclérotique bleue peut également survenir chez les nourrissons normaux au cours des premiers mois de la vie; cependant, la persistance de la décoloration bleue au fil du temps peut suggérer la présence d’une pression intraoculaire élevée. Les bébés prématurés présentent fréquemment des scléroses bleues, en particulier celles d’origine caucasienne.
La sclérotique bleue peut également apparaître isolément en tant qu’anomalie autosomique dominante ou autosomique récessive héritée 14).
Symptômes de la sclérotique bleue
La sclérotique bleue présente un aspect bleuâtre à la sclérotique et peut être associée à une cause pathologique ou non pathologique. Les autres caractéristiques oculaires des troubles du tissu conjonctif associés à la sclérotique bleue comprennent la cornée mince, le pli épicanthal, la myopie, le kératocône et les stries angioïdes. Les caractéristiques systémiques des troubles du tissu conjonctif associés à la sclérotique bleue comprennent des anomalies cutanées, des anomalies cardiaques, une cyphoscoliose, une hypermobilité articulaire, des os fragiles, des anomalies auditives, des anomalies vasculaires et des anomalies gastro-intestinales 15).
Diagnostic de la sclérotique bleue
L’évaluation diagnostique de la sclérotique bleue implique un examen externe, une biomicroscopie à lampe à fente et une évaluation systémique des troubles associés.Bien qu’il n’existe pas de test définitif pour l’ostéogenèse imparfaite, le test génétique peut confirmer ou exclure des mutations connues 16).
Traitement de la sclérotique bleue
Le traitement de la sclérotique bleue implique un diagnostic et le traitement de la cause sous-jacente. La sclérotique bleue est principalement due à des syndromes génétiques et, dans une moindre mesure, à des troubles non génétiques et peut être un effet secondaire de la prise de médicaments. Les scléroses bleues sont généralement associées à des troubles congénitaux de la synthèse du collagène, tels que l’ostéogenèse imparfaite, le syndrome de Marfan, le syndrome d’Ehlers–Danlos, le pseudoxanthome élastique et le syndrome de Willems De Vries, pour n’en nommer que quelques-uns. Les troubles osseux et sanguins figurent également sur la liste, notamment l’anémie de Diamond–Blackfan, l’anémie ferriprive sévère, la maladie de Paget juvénile et le déficit en phosphatase acide 17). Des scléroses bleues acquises ont été décrites chez des adultes atteints de mélanose oculaire, de mélanome conjonctival, d’alcaptonurie et de maladie d’Addison. Des renvois appropriés à des évaluations pédiatriques et orthopédiques peuvent être indiqués pour les manifestations non oculaires 18).
Le conseil génétique pour les troubles héréditaires associés peut être bénéfique pour les patients atteints de sclérose bleue et d’autres manifestations de maladie systémique.
Une intervention chirurgicale peut être indiquée en cas d’amincissement et de perforation extrêmes. Le support structurel peut être assuré par une greffe sclérale préservée ou un fascia lata autologue, notamment dans les cas nécessitant une suture d’un dispositif tel qu’un implant tubulaire 19). Des investigations systémiques et un traitement doivent être envisagés pour traiter la pathologie sous-jacente.
Pronostic de la sclérotique bleue
Le pronostic varie en fonction de la présence de manifestations oculaires et systémiques du trouble sous-jacent. Les patients atteints de sclérotique bleue courent un risque accru de rupture du globe ou de perforation sclérale peropératoire lors d’une chirurgie oculaire de routine. Des troubles systémiques sont également fréquents chez les patients tels que fistule carotide-caverneuse, rupture artérielle, perte auditive et fractures osseuses 20).
Références