La structure primaire d’une protéine est définie comme la séquence d’acides aminés dont elle est composée. Cette séquence détermine finalement la forme qu’adopte la protéine, en fonction des limitations spatiales de la disposition des atomes dans la protéine, des propriétés chimiques des résidus d’acides aminés constitutifs et de l’environnement de la protéine.
Les liaisons peptidiques qui lient des résidus d’acides aminés dans un polypeptide se forment dans une réaction de condensation entre le groupe carboxyle acide d’un acide aminé et le groupe amino basique d’un autre acide aminé. Dans le cadre d’un peptide, le groupe amide (CO–NH) est appelé groupe peptidique.
La connaissance de la structure de la liaison peptidique est essentielle à la compréhension de la structure des protéines. Linus Pauling, dans les années 1930, a utilisé la diffraction des rayons X pour examiner la nature de la liaison peptidique formée entre deux acides aminés. Il a rapporté que le groupe peptidique (CO–NH) a une structure plane rigide. Cette structure est due aux interactions entre les électrons de la double liaison du groupe carbonyle et ceux de la liaison C–N (Figure 2) de telle sorte que cette dernière acquiert des propriétés partielles (environ 40%) de double liaison.

Cet effet est un exemple de résonance qui peut être considéré comme un partage d’électrons entre des liaisons. Comme les liaisons simples entre deux atomes sont plus longues que les liaisons doubles entre les deux mêmes atomes, les longueurs des liaisons C–N et C = O dans le groupe peptidique diffèrent de celles observées pour ces liaisons dans d’autres contextes où la résonance ne se produit pas. Ainsi, le caractère de double liaison partielle de C-N dans le groupe peptidique signifie que cette liaison est plus courte que ce qui serait prédit pour une liaison simple C–N, tandis que la liaison C = O, ayant un caractère de liaison simple partielle dû à la résonance, est plus longue que ce qui serait prédit pour une liaison double C = O. Les longueurs de liaison dans le groupe peptidique sont indiquées à la figure 3. Comparer la liaison C-N du groupe peptidique avec celle entre N et Ca (l’atome C auquel sont attachés le groupe amino et le groupe carboxyle).

Il existe deux conformations possibles de la liaison peptidique planaire: dans le groupe peptidique trans, les atomes de Ca sont des côtés opposés de la liaison peptidique (Figure 3a) et dans le groupe peptidique cis, les atomes de Ca sont du même côté de la liaison peptidique (Figure 3b).
-
Compte tenu de la disposition spatiale et de la proximité des atomes dans les conformations cis et trans de la liaison peptidique, quelle conformation pensez-vous être privilégiée?
-
La conformation trans serait énergétiquement plus favorable que la conformation cis, car elle minimise l’obstacle stérique.
De manière générale, les liaisons peptidiques sont dans la conformation trans. Cependant, des formes cis peuvent se produire dans les liaisons peptidiques qui précèdent un résidu de proline. Dans de tels cas, la forme cis est plus stable que d’habitude puisque la chaîne latérale de proline offre moins d’obstacle. Néanmoins, les liaisons peptidiques cis ne se produisent que dans environ 10% des cas de liaisons peptidiques précédant les résidus de proline.
Compte tenu de la nature plane du groupe peptidique, on peut voir qu’une chaîne polypeptidique présente un squelette constitué d’une série de groupes peptidiques plans rigides liés par les atomes de Ca. La figure 4 montre une partie d’un polypeptide avec deux groupes peptidiques planaires dans la conformation trans. Notez que bien que la rotation ne soit pas autorisée sur les liaisons peptidiques, il existe un potentiel de rotation autour des liaisons Ca–N et Ca–C. Les angles de rotation, appelés angles de torsion, autour de ces liaisons spécifient la conformation d’un squelette polypeptidique. Les angles de torsion autour des liaisons Ca–N et Ca–C sont appelés ɸ (phi) et ψ. (psi), respectivement et ils sont définis à 180° lorsque le polypeptide est dans la conformation plane étendue, comme illustré à la figure 4.
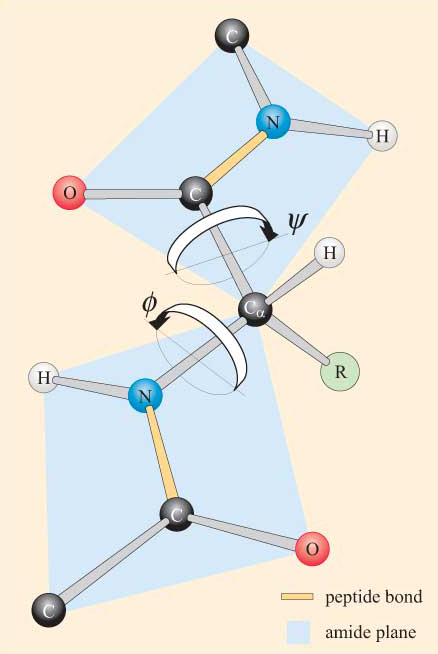
Vous ne serez pas surpris d’apprendre que des contraintes stériques s’appliquent à ψ et ψ.
Du fait de ces contraintes stériques, seules certaines valeurs de ψ et ψ, et donc des conformations du peptide, sont autorisées alors que d’autres ne le sont pas.
Il est possible de calculer ces valeurs autorisées pour un résidu donné dans le cadre d’un polypeptide. Ce calcul est effectué en déterminant d’abord les distances entre tous les atomes non liés dans deux groupes peptidiques voisins (tels que ceux de la Figure 4) à toutes les valeurs possibles de ψ et ψ. Il est plus facile de le faire pour un polypeptide contenant un seul type d’acide aminé. Un tracé conformationnel de ψ contre ψ pour un résidu particulier est connu sous le nom de tracé Ramachandran (d’après son inventeur, G. N. Ramachandran). Un tel tracé permet d’identifier les conformations (c’est-à-dire pour une valeur particulière de ψ et ψ) qui sont stériquement favorables ou défavorables (comme sur la figure 5), selon les critères suivants :
-
Lorsqu’il n’y a pas de conflit entre les rayons de van der Waals des atomes sans liaison, une conformation est « autorisée ». Ces conformations se trouvent dans les zones bleues de la figure 5.
-
Les conformations nécessitant des distances interatomiques à la limite de ce qui est admissible sont définies comme des conformations » limites extérieures « . Ils se trouvent dans les espaces verts de la figure 5.
-
Les conformations théoriques qui nécessitent que deux atomes quelconques sans liaison soient plus proches l’un de l’autre que ne le permettent leurs rayons de van der Waals sont stériquement « interdites ». Ceux-ci se trouvent dans les zones blanches de la figure 5.
Notez que les valeurs de ψ et ψ de la figure 5 vont de -180º à +180º. Tourner le groupe peptidique à 360º le ramènera bien sûr à sa position de départ, et −180º et +180º correspondent à la même position. Ainsi, la bande verte dans le coin inférieur gauche de la parcelle de la figure 5 est contiguë au champ dans le coin supérieur gauche.

-
Utilisez la figure 5 pour déterminer si les valeurs suivantes de ɸ et ψ sont stériquement favorables ou défavorables : (a) ɸ = 90º et ψ=90º; (b) ɸ=− 90º et ψ =90º.
-
(a) Défavorable; b) favorable.
Des tracés de Ramachandran peuvent être construits pour des polymères de chacun des 20 acides aminés. Il est important de noter que les parcelles de Ramachandran pour de nombreux résidus d’acides aminés sont généralement très similaires, avec seulement trois régions avec des conformations favorables ou tolérées (marquées de 1 à 3 dans la parcelle pour la poly-l-alanine de la figure 5). Des différences se produisent cependant. Par exemple, lorsque la chaîne latérale (R sur la figure 4) est ramifiée près de Ca, comme dans le cas de la thréonine, elle occupe plus d’espace près du squelette peptidique et limite l’approche des atomes dans les groupes peptidiques voisins. En conséquence, les conformations autorisées (anglesɸ et ψ) sont plus restreintes pour les polypeptides d’acides aminés ramifiés.
-
La proline est également très différente des autres acides aminés en termes de conformations autorisées et pour la polyproline seulement ɸ des valeurs de −85º à −35º sont tolérées. En pensant à la structure de la proline, comment pouvez-vous expliquer cette gamme relativement étroite de valeurs autorisées ɸ?
-
La chaîne latérale de la proline est liée de manière covalente au N du groupe amino, de sorte que dans la polyproline, il y aura moins de liberté de rotation autour de la liaison Ca-N qu’avec les autres acides aminés. Par conséquent, les valeurs autorisées ɸ seront relativement limitées par rapport aux autres acides aminés.
-
La figure 6 montre le tracé de Ramachandran pour les résidus de glycine dans une chaîne polypeptidique. Les régions sont codées par couleur comme sur la figure 5. Que pouvez-vous dire des conformations que la glycine adopte? Considérez la structure de la glycine. Pourquoi la glycine diffère-t-elle des autres résidus par ses conformations?
-
La glycine a une liberté conformationnelle beaucoup plus grande que les autres résidus d’acides aminés, car elle est moins entravée par le stéric.

Les diagrammes de Ramachandran des figures 5 et 6 ont été générés pour, respectivement, la l-alanine et la l-glycine sur la base des distances limites autorisées et externes pour les contacts interatomiques, déterminées à partir de valeurs connues pour les rayons de van der Waals des atomes (tableau 1).
Tableau 1 Distances de Van der Waals pour les contacts interatomiques.
| Type de contact | Normalement autorisé / Å | Limite extérieure / Å | |
|---|---|---|---|
| H *** H | 2.0 | 1.9 |
3.0 |
Ce sont donc des tracés conformationnels prédictifs plutôt que réels. Nous pouvons bien sûr utiliser la diffraction des rayons X pour déterminer expérimentalement les valeurs « réelles » de ψ et ψ pour les résidus d’un polypeptide. Sur la figure 7, les valeurs ɸ et ψ de tous les résidus (à l’exception de la glycine et de la proline) dans un certain nombre de structures différentes ont été déterminées par diffraction des rayons X à haute résolution et tracées sur un tracé Ramachandran. Nous pouvons voir qu’il existe une correspondance frappante entre les conformations prédites et réelles. Notez cependant qu’il existe des résidus dont les conformations correspondent aux zones « interdites ». La plupart de ces résidus cartographient dans la région entre les régions « autorisées » 2 et 3, autour de ψ = 0.

-
Regardez à nouveau la figure 4 et imaginez que vous pouvez tordre le groupe peptidique le plus élevé à 180 ° de sorte que ψ = 0. Quels groupes sont susceptibles d’entrer en conflit dans cette conformation?
-
Les groupes N-H des groupes peptidiques adjacents entreront en conflit les uns avec les autres, étant forcés de se rapprocher.
Le conflit associé à ces conformations peut être accommodé par un faible degré de torsion de la liaison peptidique. Ainsi, dans de telles conformations, le groupe peptidique est tordu hors de sa conformation planaire habituelle.
Un nombre limité de conformations « interdites » de résidus particuliers peut être toléré dans un polypeptide si la conformation adoptée, dans son ensemble, est énergétiquement favorable. Un polypeptide aura tendance à se plier de telle sorte qu’il adopte la conformation la plus stable. Dans cette conformation, le polypeptide minimise son énergie libre. Dans les sections suivantes, nous examinerons ce niveau plus élevé de structure protéique.