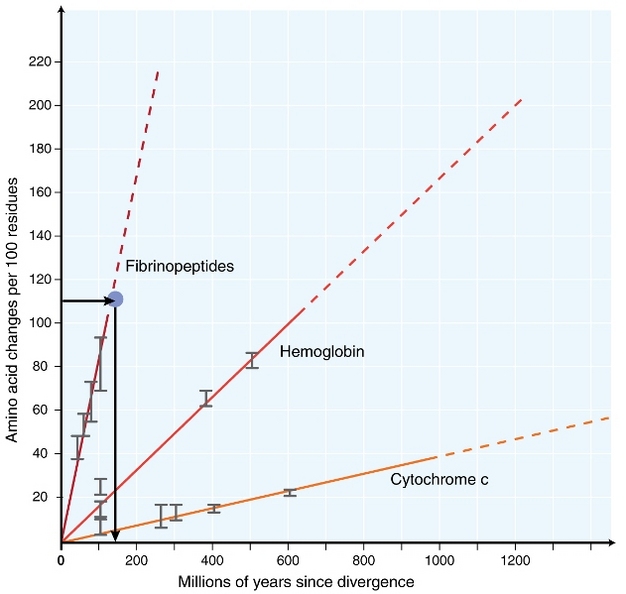
Au début des années 1960, les biologistes ont commencé à étudier comment les protéines chez différentes espèces évoluent au niveau de la séquence (Zuckerkandl 1962, Margoliash 1963, Doolittle &Blombäck 1964). Les protéines analysées comprenaient l’hémoglobine (Zuckerkandl &Pauling 1962), le cytochrome C (Margoliash 1963) et les fibrinopeptides (Doolittle&Blombäck 1964). Ces premières recherches ont conduit à une découverte remarquable : il est apparu que le nombre de différences entre les séquences protéiques de différentes espèces était à peu près proportionnel au temps écoulé depuis la divergence des espèces (Figure 1).
Zuckerkandl &Pauling (1965) a comparé l’accumulation constante de substitutions d’acides aminés au fil du temps à des « ticks » réguliers d’horloges, et a déclaré qu‘ »il peut exister une horloge évolutive moléculaire ». Ainsi, le terme « horloge moléculaire » a été initialement inventé pour décrire les changements dans les acides aminés qui se produisent proportionnellement au temps depuis la divergence des espèces.
Depuis sa première utilisation, le terme « horloge moléculaire » a été utilisé dans de nombreux contextes différents. De nos jours, il est souvent utilisé pour désigner simplement le nombre de changements, ou « substitutions », accumulés dans la séquence d’ADN ou de protéines dans une lignée donnée. Le nombre de substitutions par unité de temps définie peut être décrit comme la « vitesse » de l’horloge moléculaire, qui dans ce contexte est équivalente à la « vitesse évolutive ». Notez que la connotation initiale tient toujours dans certains cas. Par exemple, lorsque les gens déclarent ‘nous avons supposé une horloge moléculaire » (en particulier dans les analyses phylogénétiques), cela signifie que le nombre de substitutions a été supposé se produire constamment dans le temps.
Le concept d’horloge moléculaire constante a des implications extraordinaires pour les biologistes de l’évolution. Si une horloge moléculaire constante telle que proposée initialement existait vraiment, l’inférence de la synchronisation des événements évolutifs deviendrait un problème assez simple (figure 1). Cependant, il est devenu très clair que les substitutions ne se produisent pas constamment au fil du temps dans différentes lignées (Kumar 2005). Néanmoins, le concept d’horloge moléculaire a été extrêmement influent dans le domaine de l’évolution moléculaire. L’une des idées les plus importantes inspirées par le concept d’horloge moléculaire est la théorie neutre de l’évolution moléculaire.
L’Horloge moléculaire et la neutralité
Lorsque l’idée d’une horloge moléculaire constante a émergé pour la première fois, on pensait que la force évolutive prédominante sous-jacente aux substitutions d’acides aminés ou de nucléotides était la sélection naturelle. Suivant cette ligne de pensée, une horloge moléculaire constante indiquerait que des substitutions adaptatives chez différentes espèces se produisent constamment au fil du temps. Cependant, il est difficile d’expliquer comment les substitutions adaptatives se produiraient de manière aussi horloge. Théoriquement, le destin des mutations adaptatives est déterminé par plusieurs paramètres évolutifs, tels que la force de l’avantage sélectif de cette mutation, la taille de la population effective et les taux de mutation adaptative (Kimura 1983). Ces paramètres sont susceptibles de différer d’une espèce à l’autre, et même au sein d’une espèce, en fonction de mutations spécifiques et de leurs interactions avec les environnements.
Au lieu de cela, Kimura (1968, 1969) a proposé que la plupart des changements au niveau moléculaire ont peu de conséquences fonctionnelles, ou sont « neutres ». Si une mutation n’a aucune conséquence sur la condition physique, son sort dans la population est déterminé complètement par hasard. Cela signifie que nous ne pouvons pas prédire si une mutation neutre spécifique sera éventuellement fixée dans la population. Cependant, le taux auquel des substitutions neutres se produisent dans la population peut être prédit car il dépend d’un seul paramètre, à savoir le taux de mutation (Kimura 1968).
Imaginons une population avec N nombre d’individus haploïdes. Si des mutations neutres se produisent au taux u par individu et par génération, le nombre total de mutations dans une génération sera N fois u. Puisque toutes ces nouvelles mutations sont neutres, leur destin est complètement déterminé par hasard. En d’autres termes, toutes les mutations ont des chances égales d’atteindre la fixation (ce qui conduit à une « substitution »). La probabilité que chaque nouvelle mutation neutre atteigne la fixation, étant donné qu’une substitution s’est produite, est simplement de 1/ N. Le taux de substitutions est calculé comme le nombre de nouvelles mutations à chaque génération (Nu) multiplié par la probabilité que chaque nouvelle mutation atteigne la fixation (1/N), ce qui équivaut à u. En d’autres termes, pour les mutations neutres, le taux de substitution est égal au taux de mutation!
Par conséquent, si la plupart des mutations sont neutres (comme proposé dans la théorie neutre) et si les taux de mutation sont constants dans le temps, les substitutions devraient également se produire constamment dans le temps. Nous devrions alors observer des taux réguliers de substitutions au niveau moléculaire. Kimura (1969) a donc considéré l’observation d’une horloge moléculaire relativement constante dans les séquences protéiques comme un support solide pour la théorie neutre de l’évolution moléculaire.
Tester le fonctionnement de l’Horloge moléculaire neutre
Selon la théorie neutre, la question de savoir si les taux de substitution sont constants dans le temps ou non équivaut à savoir si les taux de mutation neutres sont constants dans le temps. Pour cette raison, de nombreuses études ultérieures se sont concentrées sur l’analyse de données provenant de sites neutres afin de déterminer si les taux de mutation neutres sont effectivement constants dans le temps. Nous passerons brièvement en revue la manière dont ces études sont mises en œuvre, avant de discuter des débats théoriques sur les horloges moléculaires. Les analyses des horloges moléculaires des protéines se sont également poursuivies, mais les débats entourant la variation des horloges moléculaires des protéines sont très différents de ceux concernant les horloges moléculaires neutres et ne seront pas inclus dans cet article. Les lecteurs intéressés devraient consulter Gillespie (1991), Kumar (2005), Kim et Yi (2008), et Bedford et al. (2008).
La plupart des analyses empiriques des horloges moléculaires neutres reposent sur le théorème selon lequel les taux de mutation neutres peuvent être déduits des taux de substitution neutres (Kimura 1968, 1969). En pratique, chaque étude définit un certain type de sites du génome comme des sites neutres et compare les taux de substitution de ces sites entre lignées.
Les sites du génome qui sont vraiment neutres ne peuvent pas être complètement déterminés, mais les scientifiques ont mis au point plusieurs procurations utiles. Avant l’ère du séquençage du génome, la plupart des données de séquence disponibles étaient celles de séquences d’ADN codant des protéines. Les études divisent souvent les séquences d’ADN codant pour les protéines en deux types de sites (Wu&Li 1985). Le premier type de sites comprend ceux pour lesquels tout changement entraînerait des substitutions d’acides aminés, ou « sites non synonymes ». Le deuxième type de sites, les « sites synonymes », comprend ceux qui codent des positions « dégénérées » dans la table de codon, où un changement n’entraîne pas de substitution d’acides aminés. Par exemple, TCT et TCC codent tous deux la sérine. Si la troisième position de ces codons change, ils produiront toujours les mêmes acides aminés. De telles substitutions seraient moins visibles pour la sélection naturelle. Par conséquent, les horloges moléculaires des sites synonymes devraient être plus proches de l’horloge moléculaire neutre que les horloges non synonymes.
Au fur et à mesure que les techniques de séquençage progressaient, certaines études ont utilisé des introns comme substituts pour des sites neutres, car ils ne sont pas incorporés dans les ARNM matures et sont donc plus susceptibles d’être neutres (Yi et al. 2002). Des séquences d’éléments transposables inactifs qui ont été insérés bien avant la divergence des espèces ont également souvent été utilisées (on parle souvent de » répétitions ancestrales « , par exemple, Thomas et al., 2003). Enfin, certaines études ont utilisé des séquences d’ADN non codantes (toutes les séquences après élimination des séquences d’ADN codant des protéines) extraites d’alignements de génome entier pour tester des horloges moléculaires neutres (Elango et al. 2006, Huttley et coll. 2007).
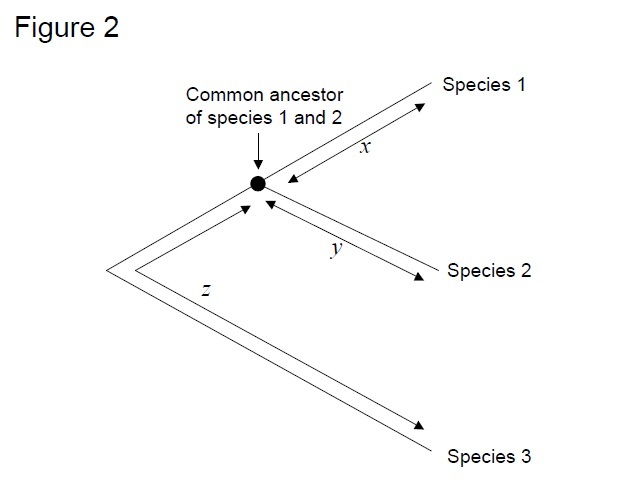
Le test le plus couramment utilisé est le « test de taux relatif » (Sarich &Wilson 1973). Initialement, les taux de substitution par unité de temps ont été estimés en divisant le nombre total de différences (substitutions) entre les protéines de différentes espèces par le temps de divergence, estimé à partir des enregistrements fossiles (figure 1). Cependant, les enregistrements fossiles ne sont pas disponibles pour de nombreuses comparaisons et sont associés à de grandes marges d’erreur. Le test du taux relatif permet de surmonter la nécessité d’enregistrer des fossiles (figure 2). Tant qu’il existe une séquence de groupes externes aux deux lignées d’intérêt, nous pouvons déterminer si les deux branches suivent les mêmes horloges moléculaires ou des horloges moléculaires différentes, sans connaître le temps absolu de divergence (Figure 2).
Quels sont les Déterminants des Horloges moléculaires neutres?
Presque toutes les controverses au cœur des débats sur les horloges moléculaires neutres proviennent de la question de savoir quelles sont les principales sources de mutations. Cette question est directement pertinente pour comprendre les modèles de mutation, qui sont la source ultime du changement évolutif et des maladies génétiques. De plus, comprendre comment les taux de mutation varient entre les lignées et au sein des génomes est une question fondamentale en génomique comparative, qui vise à utiliser des comparaisons de séquences pour identifier les régions génomiques qui sont fonctionnellement importantes.
Qu’est-ce qui détermine les taux de mutation neutres ? L’un des contributeurs les plus importants aux horloges moléculaires neutres est la variation spécifique à la lignée dans les temps de génération. Dès le début, l’idée d’une horloge moléculaire neutre constante a été perçue comme étant en contradiction avec les mécanismes moléculaires de la mutation germinale. On a longtemps considéré que la plupart des mutations résultent d’erreurs de réplication de l’ADN dans les lignées germinales (Haldane 1947, Muller 1954). Étant donné que les mutations se produisent lorsque l’ADN de la lignée germinale est répliqué pour la génération suivante, elles devraient s’accumuler proportionnellement au nombre de générations, plutôt qu’au temps absolu. Par conséquent, si nous comparons le nombre de substitutions qui se sont accumulées dans deux lignées depuis leur divergence, la lignée avec un temps de génération plus long, ayant subi moins d’événements de réplication de l’ADN, abriterait moins de substitutions par rapport à la lignée avec le temps de génération plus court. Par conséquent, l’horloge moléculaire devrait fonctionner plus lentement chez les espèces avec des temps de génération plus longs. Cette idée est appelée « effet génération-temps ».
En fait, l’effet génération-temps a été observé pour la première fois dans des études sur des primates, avant même le débat sur l’horloge moléculaire. Morris Goodman, qui utilisait des méthodes immunologiques pour étudier la parenté entre espèces à l’époque, a observé que la vitesse à laquelle certaines protéines divergent semblait être diminuée chez les singes, en particulier les humains, par rapport aux singes de l’Ancien Monde (Goodman 1961, 1962, 1963). Cet effet est appelé « ralentissement du taux d’hominoïdes ». Comme les hominoïdes ont des temps de génération plus longs que les singes de l’Ancien Monde, cette observation peut s’expliquer par l’effet génération-temps.
Wu &Li (1985) ont été les premiers à tester l’hypothèse de l’effet de génération en utilisant des données de séquence d’ADN. Ils ont utilisé des données provenant de 11 gènes de primates et de rongeurs. Comme les primates ont un temps de génération beaucoup plus long que les rongeurs, l’horloge moléculaire devrait être plus rapide chez les rongeurs que chez les primates. En effet, ils ont constaté que pour les sites synonymes, les rongeurs présentent environ deux fois le taux d’évolution moléculaire par rapport aux primates (Wu&Li 1985). Cependant, pour les sites non synonymes, un tel effet n’a pas été trouvé. En d’autres termes, l’horloge moléculaire neutre, mais pas l’horloge moléculaire des acides aminés, tique plus rapidement dans la lignée des rongeurs par rapport à la lignée des primates, ce qui correspond bien à l’idée d’un effet de génération.
Des études ultérieures ont apporté un soutien supplémentaire au ralentissement du taux d’hominoïdes (Li&Tanimura 1987, Bailey et al. 1991) et la différence de taux entre les lignées de rongeurs et de primates (Gu &Li 1992, Huttley et al. 2007). De plus, une différence de taux a été observée à des échelles phylogénétiques encore plus petites, en particulier chez les primates: par exemple, l’horloge moléculaire humaine tourne plus lentement que l’horloge moléculaire du chimpanzé (Elango et al. 2006); les taux chez les singes du Nouveau Monde sont plus rapides que chez les hominoïdes et les singes de l’Ancien Monde (Steiper &Young 2006). Les différents taux d’horloges moléculaires observés dans ces études sont qualitativement compatibles avec l’effet génération-temps.
Cependant, les différences réelles entre les lignées ne sont pas quantitativement cohérentes avec la différence de temps de génération. Par exemple, Kumar &Subramanian (2002) a montré que même si la différence de temps de génération entre les primates et les rongeurs est beaucoup plus grande que celle entre les humains et les singes de l’Ancien Monde, les différences observées dans les horloges moléculaires sont similaires dans ces deux comparaisons. Il est intéressant de noter que Kumar &Subramanian (2002) a utilisé des filtres statistiques spécifiques pour supprimer les données montrant des modèles de substitution « hétérogènes », ce qui aurait pu provoquer un biais vers des séquences évoluant lentement (Yi et al. 2002). Néanmoins, la différence dans les horloges moléculaires des primates et des rongeurs semble beaucoup moins importante que celle proposée initialement par Wu &Li (1985). Par exemple, Huttley et al. (2007) ont analysé les alignements du génome entier de plusieurs espèces, y compris l’opossum, et ont montré que la différence de taux entre les lignées euthériennes et la lignée opossum (~ 30%) est beaucoup plus grande que la différence de taux entre les lignées humaines et murines (~ 14%). Ces exemples démontrent que le degré de différences dans les horloges moléculaires varie considérablement d’une étude à l’autre en raison de différences dans les ensembles de données et les méthodes statistiques. Ils montrent également que les différences de taux entre les lignées ne peuvent être entièrement expliquées par la seule différence de temps de génération. De toute évidence, il existe d’autres contributeurs aux horloges moléculaires neutres.
En effet, les traits de l’histoire de la vie autres que les temps de génération semblent co-varier avec les horloges moléculaires. Martin &Palumbi (1993) a montré que les horloges moléculaires de l’ADN fonctionnent plus rapidement chez les espèces de petite taille. Cette observation a conduit à l’hypothèse que les taux métaboliques sont des déterminants importants des horloges moléculaires. Un taux métabolique élevé produit un grand nombre de radicaux oxygénés mutagènes, ce qui augmenterait les taux de mutation (Rand, 1994). Étant donné que les taux métaboliques et la taille du corps varient généralement en fonction des temps de génération, il a été difficile de distinguer lequel d’entre eux constitue le principal déterminant des cadences d’horloge moléculaire. Les Tsantes &Steiper (2009) ont proposé, sur la base de données de primates, que l’âge à la première reproduction, plutôt que la taille du corps, est le principal déterminant des horloges moléculaires. Puisque l’âge à la première reproduction reflète l’effet du temps de génération, cette étude soutient l’idée que l’effet du temps de génération est le principal déterminant de l’horloge moléculaire. Cependant, cette étude est toujours basée sur un nombre limité de lignées (quatre paires d’espèces ont été utilisées). Ainsi, la distinction entre la taille du corps, l’effet de génération et les taux métaboliques reste un problème important pour généraliser et comprendre les horloges moléculaires neutres.
En outre, l’importance des facteurs qui ne co-varient pas avec les temps de génération, tels que la méthylation de l’ADN, a été de plus en plus appréciée. La méthylation de l’ADN est une modification chimique de l’ADN génomique présente dans divers taxons. Dans les génomes animaux, la méthylation de l’ADN se produit presque exclusivement au niveau des cytosines suivies des guanines (appelées « CpG »). Les cytosines méthylées, quant à elles, ont tendance à muter rapidement en thymines en raison de l’instabilité chimique (Bird, 1980). En effet, dans le génome humain, les mutations causées par la méthylation de l’ADN se produisent plus d’un ordre de grandeur plus fréquemment que d’autres mutations (Nachman &Crowell 2000, Elango et al. 2008). Étant donné que les mutations causées par la méthylation de l’ADN se produisent en grande partie indépendamment de la réplication de l’ADN, de telles mutations peuvent suivre des horloges moléculaires différentes des autres. Plus précisément, au lieu de la dépendance au temps de génération, les mutations causées par la méthylation de l’ADN peuvent suivre une horloge moléculaire dépendante du temps, ce qui est similaire à ce qui a été initialement proposé par Zuckerkandl &Pauling (Kim et al. 2006)!
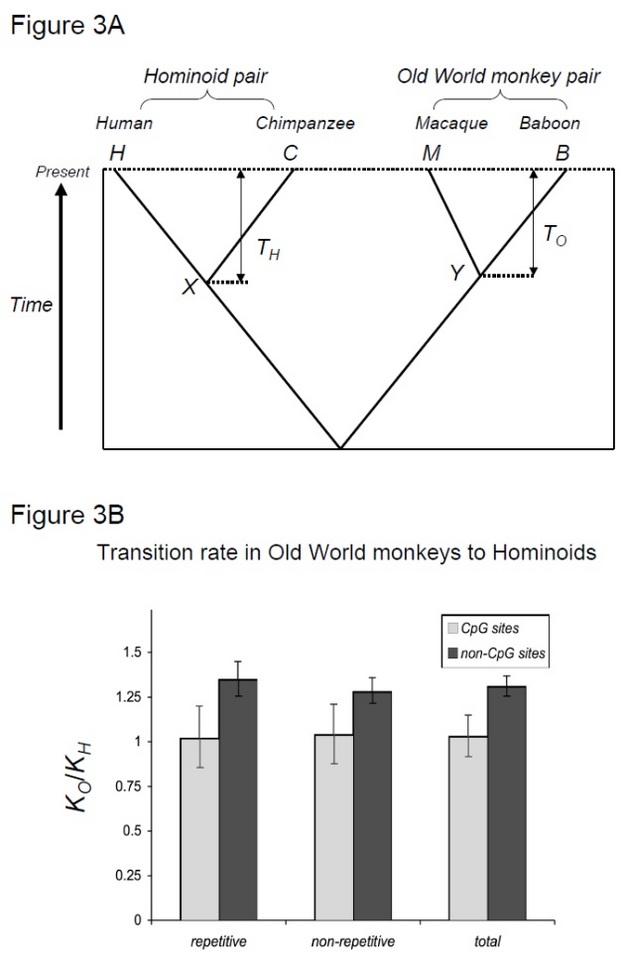
Pour tester cette hypothèse, Kim et al. (2006) ont comparé la divergence homme-chimpanzé à la divergence macaque-babouin, deux paires d’espèces qui partagent des temps de divergence similaires mais avec des temps de génération différents (Steiper et al. 2004). La paire homme-chimpanzé (la paire hominoïde) a des temps de génération beaucoup plus longs que la paire macaque-babouin (la paire singe de l’Ancien Monde). Cette étude a montré que pour les sites non CpG, le couple de singes de l’Ancien Monde a accumulé environ 30% de substitutions supplémentaires, ce qui peut s’expliquer par l’effet de ralentissement du taux d’hominoïdes susmentionné. En revanche, les horloges moléculaires des sites CpG ont montré un nombre similaire de substitutions dans les paires d’hominoïdes et de singes de l’Ancien Monde (Figure 3). Ainsi, des horloges moléculaires dépendantes du temps et du temps de génération coexistent dans les mêmes génomes. L’hypothèse selon laquelle une seule horloge moléculaire peut exister pour une lignée donnée n’est plus valable, car les forces mutationnelles prédominantes varient d’une région génomique à l’autre.
Conclusions
Le concept d’horloge moléculaire constante a été initialement proposé sur la base d’une quantité limitée de données de séquence de protéines. Même si des études ultérieures ont montré qu’une telle observation n’est pas un modèle général dans les acides aminés, elle a eu une influence significative sur le domaine de l’évolution moléculaire, en particulier sur le développement de la théorie neutre de l’évolution moléculaire pour les données de séquence d’ADN. Suivant la théorie neutre, les études se sont concentrées sur l’élucidation des modèles de variation des taux de mutation neutres. Au cours des dernières décennies, nous avons observé que les horloges moléculaires fonctionnent à des vitesses différentes entre les lignées. De plus, le degré de variation peut varier en fonction des différents types de données et des méthodes statistiques spécifiques utilisées. L’effet génération-temps continue de se maintenir à un niveau qualitatif, mais est insuffisant pour expliquer la variation quantitative des taux de mutation neutres entre les lignées. Les traits du cycle de vie et les mutations non dépendantes de la réplication, telles que celles causées par la méthylation de l’ADN, sont également des contributeurs importants aux horloges moléculaires génomiques. En effet, différents types d’horloges moléculaires sont observés même au sein d’un génome, car les entrées mutationnelles prédominantes varient entre les différentes régions génomiques. Ainsi, plutôt que de supposer une horloge moléculaire neutre unique pour chaque génome, les futures études devraient viser à révéler la variation des horloges moléculaires neutres génomiques, pour en apprendre davantage sur les paysages mutationnels génomiques. Ces informations sont non seulement utiles pour comprendre la matière première régissant l’évolution moléculaire et les maladies génétiques, mais constituent également un élément essentiel influençant les analyses génomiques comparatives et fonctionnelles pour identifier les régions génomiques fonctionnelles.