- Définitions et prévalence
- Diagnostic
- Manifestations du CCNSA chez la femme
- Manifestations dans l’enfance
- Manifestations à l’adolescence et à l’âge adulte
- Prise en charge du CCNSA De la première manifestation à la vie adulte
- Prise en charge Pendant l’enfance
- Prise en charge Pendant l’adolescence
- Patientes Traitées Depuis l’enfance
- Femmes Présentant Une Première Manifestation Pendant l’Adolescence ou Des Symptômes Hyperandrogènes Après l’arrêt du traitement
- NCCAH et reproduction
- Subfertilité
- Fausses couches
- Planification de la fertilité
- Traitement prénatal de la CAH
- Traitement Pendant la grossesse
- Autres Problèmes spéciaux
- Gestion du stress dans le CCNSA
- Aspects psychobiologiques dans le CCNSA
- Conclusions
- Contributions des auteurs
- Déclaration sur les conflits d’intérêts
Définitions et prévalence
L’hyperplasie surrénalienne congénitale (CAH) englobe une famille de troubles autosomiques récessifs caractérisés par une altération légère à aiguë de la synthèse du cortisol due à une déficience de l’une des cinq enzymes stéroïdogènes surrénaliennes nécessaires à la production de cortisol (1, 2). Classiquement, la CAH est divisée en (a) classique (CCAH), se présentant sous une forme de salinisation ou de simple virilisation qui se manifeste à la naissance et / ou au cours de la période néonatale, et (b) non classique (NCCAH), représentant une forme moins grave du trouble qui ne présente pas d’ambiguïté génitale, ne met pas immédiatement la vie en danger et se présente plus tard dans la vie, ou reste asymptomatique, ou est mal diagnostiquée comme une maladie différente (3). Cependant, les limites entre les différentes formes de CAH ne sont pas strictement définies, ce qui tend à augmenter les défis associés à ce trouble. Il est donc conseillé de considérer la CAH comme un continuum de phénotypes, de sévère à léger ou bien asymptomatique (3).
La forme la plus courante de CAH est causée par un déficit en 21-hydroxylase (21OHD), entraînant une altération ou l’absence de conversion de 17 hydroxyprogestérone (17 OHP) en 11 désoxycortisol et de la progestérone en désoxycorticostérone. Le blocage de la conversion des stéroïdes entraîne une augmentation de la production de précurseurs d’androgènes, sous stimulation CRH-ACTH, conduisant à un hyperandrogénie biochimique, marqué par des niveaux élevés de 17 OHP. La forme classique de la maladie survient dans 1 naissance vivante sur 16 000 dans le monde. Cependant, il convient de mentionner que l’incidence perçue de la maladie est liée à la méthode de dépistage utilisée. Par exemple, il a été rapporté que l’incidence de la forme classique de CAH en Suède est de 1: 11 500 lorsqu’une approche d’enquête de cas est utilisée, chiffre qui tombe cependant à 1: 9 800 lorsque le dépistage hormonal est appliqué. De même, l’incidence en France et en Suisse varie entre 1:15 472 et 1:23 000 et 1:10 749 et 1:11 661 lorsque les différentes méthodes de dépistage sont utilisées (4).
Le NCCAH est beaucoup plus fréquent, se produisant chez environ 1 Caucasien sur 1 000 et plus fréquemment dans certains groupes ethniques, tels que les Juifs ashkénazes (1:27), les Hispaniques (1:53), les Yougoslaves (1:62) et les Italiens (1:300) (5, 6). Néanmoins, les données sur la prévalence du CCNSA basées sur différentes méthodes d’estimation (enquête sur les cas, dépistage hormonal et tests moléculaires) font défaut. Le NCCAH est considéré comme le trouble endocrinien autosomique récessif le plus courant avec une fréquence porteuse de 1: 25 à 1: 10. Dans le NCCAH en raison de la 21-OHD, l’activité enzymatique résiduelle est estimée à environ 10-70 % (7-9).
Diagnostic
Par rapport au diagnostic de la forme classique de la maladie, qui est posé à la naissance ou pendant la période néonatale en raison d’une ambiguïté génitale et / ou de symptômes de gaspillage de sel ou par le biais de programmes de dépistage utilisés dans certains pays, la plupart des cas de CCNSA ne sont pas facilement détectables (4, 10). De plus, de nombreuses personnes demeurent asymptomatiques pendant l’enfance et l’adolescence, ont une fonction de reproduction normale et ne prennent conscience du CCNSA qu’en raison du diagnostic d’un autre membre de la famille et des tests qui en découlent (11). Cependant, la plupart des femmes atteintes du CCNSA demandent de l’aide médicale lorsqu’elles présentent des symptômes d’excès d’androgènes et, lorsque la suspicion clinique incite à effectuer des tests, des niveaux basaux élevés de 17 OHP indiqueront plus probablement qu’autrement un diagnostic de CCNSA.
En effet, les lignes directrices cliniques proposées par l’Endocrine Society recommandent une valeur de base non stimulée de 17 OHP comme outil de dépistage du CCNSA. Les niveaux d’OHP du matin 17 > 6 nmol / L dans la phase folliculaire chez les femmes menstruées devraient inciter à une évaluation plus approfondie, car il a été démontré que des niveaux supérieurs à 6 nmol /L capturent 90% des individus du CCNAA (12, 13). Les mesures aléatoires de 17 OHP ne se sont pas révélées utiles, car elles donnent souvent des taux normaux chez les patients atteints de NCCAH, et elles sont de plus extrêmement élevées dans la phase lutéale chez la moitié des femmes en bonne santé (13). Cependant, dans nos données dérivées de 280 sujets atteints de la maladie, six patients (2.1%) avait une valeur de base de 17 OHP < 6 nmol/L (14). De plus, Bidet et coll., dans une grande cohorte de femmes atteintes de NCCAH vérifiées par des techniques moléculaires, ont également trouvé des valeurs basales de 17 OHP inférieures à 6 nmol / L chez 8% des sujets étudiés (15). Enfin, sur la base des données recueillies par Speiser et al., 9 % des personnes atteintes de NCCAH affichaient 17 valeurs d’OHP inférieures à 2 ng/ml (ce qui correspond à 6nmol/L). Selon d’autres études, une valeur de base de 17 OHP comprise entre 5,1 et 9 nmol /L est suffisante pour le diagnostic de NCCAH (13, 16, 17). Récemment, un niveau de 17 OHP basal de 4.6 nmol/L a été suggéré comme seuil pour le test ACTH pour prédire le NCCAH chez les sujets atteints d’adrénarche prématurée pendant l’enfance (18).
La somme de ces résultats et suggestions indique que la sélection des patients qui subiront un test de stimulation de la Synacthène doit être évaluée au cas par cas. Un niveau de post-stimulation de 17 OHP supérieur à 3 nmol/L est requis pour le diagnostic (19). Les hétérozygotes porteurs d’une mutation du CYP21A2 présentent des niveaux légèrement élevés de 17 OHP après la stimulation par l’ACTH, bien qu’il y ait un chevauchement chez les sujets non affectés (9). Cependant, comme Dacou-Voutetakis et al. ont souligné, si la somme des valeurs de 17 OHP basales et post-stimulation dépasse 1,5 nmol/L, alors la possibilité d’hétérozygotie est exceptionnellement élevée (20).
Une autre considération importante concerne les techniques utilisées pour l’évaluation de 17 OHP. 17 Niveaux d’OHP sont mesurés par diverses méthodes d’immunoessai, mais comme cela a été récemment montré, les résultats les plus précis et les plus fiables ont été obtenus par la mise en œuvre de la combinaison de chromatographie liquide avec spectrométrie de masse (LC-MS / MS). En effet, de nombreuses mesures faussement positives de 17 OHP ont été trouvées lorsque les mesures LC-MS / Ms ont été comparées aux méthodes standard (21) Néanmoins, ces dernières procédures ne sont pas universellement utilisées, en cas de diagnostic incertain, elles fourniront des résultats plus précis. Il est à noter que les seuils de dépistage et de diagnostic pour la LC-MS / MS quantifiée 17 OHP restent à définir. Ces seuils sont susceptibles d’être inférieurs à ceux établis avec les immunoessais en raison de la spécificité accrue de la LC-MS/ MS, une méthode moins sujette à la réactivité croisée et aux interférences (22, 23). La question de savoir si un profil de stéroïdes urinaires est nécessaire pour le diagnostic définitif reste à élucider (22). Dans les cas limites, il est conseillé d’obtenir un profil corticosurrénal complet après le test de stimulation de l’ACTH pour différencier le déficit en 21-hydroxylase des autres défauts enzymatiques et établir un diagnostic ferme.
Plus précisément, un profil de stéroïdes complet doit être effectué dans des cas équivoques pour confirmer un déficit en 21-hydroxylase et exclure d’autres détectations enzymatiques. L’inclusion de 17 OHP, de cortisol, de 11-désoxycorticostérone, de 11-désoxycortisol, de 17-OH-prégnénolone, de déhydroépiandrostérone et d’androstènedione dans un profil de stéroïdes sériques serait utile pour exclure d’autres causes de CAH telles que le déficit en 11β-hydroxylase et, plus rarement, le déficit en 3β-hydroxystéroïde déshydrogénase ou le déficit en P450 oxydoréductase (24). Bien que les tests génétiques ne soient pas considérés comme un outil de diagnostic principal pour le CCNSA à l’heure actuelle, ils sont obligatoires pour la confirmation du diagnostic et pour le counseling génétique (3).
En ce qui concerne les taux de cortisol, on s’attend généralement à une valeur post-stimulation d’au moins 496 nmol/L. À noter, selon Stoupa et al., 60% des 47 enfants atteints de NCCAH à la suite de 21 OHD avaient de faibles valeurs de cortisol après la stimulation, une constatation indiquant la nécessité d’une surveillance accrue du développement d’une insuffisance surrénale lors d’événements stressants majeurs (25).
Manifestations du CCNSA chez la femme
L’expression clinique du CCNSA se caractérise par un niveau élevé de polymorphisme en ce qui concerne non seulement l’âge d’apparition, mais également les différents signes et symptômes. Il est rapporté que la première présentation clinique du CCNSA se produit dans 11 % des cas avant l’âge de 10 ans et dans 80 % des cas entre 10 et 40 ans (12). La corrélation génotype-phénotype dans le CAH et le NCCAH n’a pas encore été élucidée. Speiser et coll. suggèrent que la plus grande partie, mais pas la totalité, de la variabilité phénotypique du déficit en 21-hydroxylase résulte de la variation allélique du CYP21A2(26).
Manifestations dans l’enfance
Pendant la période du nouveau-né, les femelles du NCCAH restent généralement asymptomatiques et ont des organes génitaux externes normaux. Le premier cas de CCNSA signalé est une fille de 6 mois qui a développé des poils pubiens (11). Habituellement, les résultats cliniques et les symptômes dans les cas de CCNSA commencent à partir de l’âge de 5 ans ou même plus tard et sont liés à une augmentation des taux d’androgènes, bien qu’une légère carence en cortisol puisse également survenir dans certains cas (8). Dans 92% des cas de CCNSA, le premier symptôme qui se manifeste est la puberté prématurée (PP), qui survient à moins de 8 ans chez les filles et à moins de 9 ans chez les garçons (12) avec une prévalence estimée entre 10 et 11,3% (15). Il est à noter que chez les enfants référés avec PP, selon différentes études, l’incidence du CCNSA varie de 5 à 30% (20, 27). Cependant, sur la base de notre analyse de 280 patients avec confirmation moléculaire du NCCAH, nous avons constaté que l’incidence de PP chez 94 femmes de moins de 8 ans était de 88%. D’autre part, une analyse de 45 hommes atteints du CCNSA a permis d’identifier la PP chez seulement 29 % des sujets (14). De plus, nous devons garder à l’esprit qu’environ la moitié des sujets atteints de PP peuvent être porteurs hétérozygotes d’une mutation du CYP21A2.
Un autre aspect qui doit être clarifié est la relation entre la PP et la puberté précoce. On suppose que dans certains cas, l’aromatisation périphérique des androgènes surrénaliens en œstrogènes peut activer l’axe hypothalamo-hypophyso-gonadique, conduisant à une puberté précoce secondaire (28). Dans un tel cas, un suivi régulier est nécessaire. Certains enfants développent de l’hirsutisme, de l’acné et/ou une croissance rapide et manifestent une structure haute et un âge osseux avancé (29). Ce dernier peut entraîner une hauteur finale tronquée à la suite d’une fusion épiphysaire rapide. D’autres résultats cliniques rares pendant l’enfance incluent l’adhésion labiale, les poils périanaux, la clitoromégalie et l’enrouement de la voix (30-32).
Manifestations à l’adolescence et à l’âge adulte
À l’adolescence et à l’âge adulte, le NCCAH chez les femmes présente des symptômes hyperandrogènes ressemblant au syndrome des ovaires polykystiques (SOPK) (33). Les symptômes comprennent l’hirsutisme, l’acné, l’alopécie androgénique, l’anovulation, le dysfonctionnement menstruel et l’infertilité. Dans une étude multicentrique, les symptômes les plus courants chez les adolescentes et les femmes adultes étaient l’hirsutisme (59%), l’oligoménorrhée (54%) et l’acné (33%). Parmi les 161 femmes atteintes de NCCAH, les symptômes présentés étaient l’hirsutisme (78 %), un dysfonctionnement menstruel (54,7 %) et une diminution de la fertilité (12 %) (34). Dans notre groupe de 161 femmes atteintes de la maladie, 63% des patientes présentaient un phénotype de type ovaire polykystique (14). Cette découverte a une implication inverse, puisque l’incidence de NCCAH est élevée chez les femmes atteintes d’hyperandrogénémie. Certes, l’étude de Witcel et al. il est très intéressant de signaler une incidence du CCNSA variant de 1 à 33 % chez les femmes hyperandrogènes. Dans une autre étude portant sur 950 femmes référées pour une évaluation de l’excès d’androgènes, le NCCAH a été détecté chez 4,2% d’entre elles. Cependant, les patients du CCNSA étaient plus jeunes et plus hirsutes que les autres groupes de patients hyperandrogènes. Ils ont également été caractérisés par des niveaux significativement plus élevés de testostérone, de testostérone libre et de 17 OHP que les patients atteints d’autres syndromes hyperandrogènes. Dans l’échographie ovarienne, 77% d’entre eux ont montré des ovaires polykystiques et 41% ont augmenté la taille des ovaires. En résumé, ils remplissaient les critères du SOPK selon NIH et Rotterdam à un pourcentage de 56 et 72,8%, respectivement, (35).
La nature progressive de la maladie est mise en évidence par le fait qu’il a été démontré que la prévalence de l’hirsutisme augmentait avec l’âge et qu’elle était rare avant la puberté. L’hirsutisme est la manifestation clinique la plus fréquente rapportée chez les patients atteints de NCCAH, allant de 71 à 96 % (28, 36). Les filles pubertaires atteintes du CCNSA présentent généralement un hirsutisme (37, 38). À l’autre extrémité du spectre se trouve la question de l’alopécie. Une calvitie masculine est rapportée chez les patients atteints du CCNSA. La prévalence de l’alopécie semble également augmenter avec l’âge, passant de 6% chez les patients au cours de la deuxième décennie de leur vie à 19% au cours de la cinquième, indiquant à nouveau la nature progressive de la maladie (39). L’acné est rapportée chez près de 33 % des sujets du CCNSA (36). Remarquablement, des mutations du gène CYP21A2 causant le NCCAH ont été détectées chez 4,9 % des 123 femmes adultes présentant une acné sévère (40).
Les irrégularités menstruelles, y compris l’oligoménorrhée ou l’aménorrhée secondaire, peuvent souvent être le signe présent du CCNSA chez les individus post-ménarche. De plus, 8 à 9% des cas présentent également une aménorrhée primaire et, pour cette raison, consultent un médecin pour la première fois. De plus, chez les individus du CCNSA, l’oligoménorrhée est plus fréquente que l’aménorrhée primaire. Par exemple, dans une série de Moran et al. chez les femmes du CCNSA âgées de 10 à 19 ans, l’oligoménorrhée était présente dans 56 % des cas, comparativement à seulement 9 % des adolescentes atteintes d’aménorrhée primaire (12). Pour conclure, tant de symptômes du CCNSA ressemblent à ceux du SOPK qu’il n’est pas surprenant que le CCNSA ait été surnommé le grand imitateur du SOPK. Dans tous les cas, un diagnostic de CCNSA doit être envisagé lors de l’évaluation de toute jeune femme référée pour des symptômes hyperandrogènes.
Dans une étude réalisée par Arlt et al., les patients atteints de NCCAH ont été caractérisés par un IMC significativement plus élevé et une hauteur finale inférieure par rapport aux témoins appariés (41). Ces données, associées à quelques études qui indiquent la présence d’une insensibilité à l’insuline, suggèrent un profil métabolique défavorable qui pourrait s’expliquer physiopathologiquement par l’effet de concentrations élevées d’androgènes et / ou à la suite d’un traitement aux glucocorticoïdes (42, 43). L’ostéoporose peut également être détectée chez ces sujets, probablement à la suite du traitement par corticostéroïdes (34).
Prise en charge du CCNSA De la première manifestation à la vie adulte
Une fois le diagnostic du CCNSA établi, plusieurs questions liées aux recommandations de traitement ultérieures méritent d’être prises en compte. La première question à aborder est de savoir si un traitement aux glucocorticoïdes (GC) est indiqué. Le raisonnement général derrière cette approche est qu’en fournissant des concentrations suffisantes de cortisol au patient, ses besoins quotidiens sont couverts et, par conséquent, la stimulation de l’axe CRH-ACTH sera effilée, entraînant une diminution de la production d’androgènes surrénaliens. La règle générale concernant cette approche est que les GCs sont administrés à des doses de remplacement et non pharmacologiques, tandis que l’influence de l’âge, du sexe, des données de laboratoire, des recommandations spécifiques au patient et des objectifs du traitement sur le traitement de remplacement des glucocorticoïdes est sérieusement prise en compte. Cependant, nous devons également être conscients que la thérapie de substitution GC prolongée peut entraîner une hypercortisolémie, avec les effets indésirables bien documentés qui en résultent sur tous les aspects du métabolisme, en particulier la croissance, la distribution des graisses et la résistance à l’insuline ainsi que sur le profil psychologique. Un autre inconvénient majeur de cette approche est l’absence d’un indice clinique adéquat ou d’un marqueur biochimique de dosage de remplacement adéquat, tel qu’il existe en ce qui concerne les valeurs de TSH dans l’hypothyroïdie.
Enfin, le fait qu’il n’y ait pas de consensus sur la GC à utiliser et l’absence de données à long terme concernant les différentes modalités d’administration de la GC compliquent encore davantage les décisions des médecins traitants et le choix du choix optimal pour chaque patient. L’hydrocortisone est généralement utilisée chez les enfants, car elle ressemble le plus à l’hormone naturelle (cortisol), mais elle n’est pas considérée comme une approche appropriée chez les adolescents et les jeunes femmes en raison de la nécessité de doses quotidiennes multiples. Par conséquent, la plupart des endocrinologues adultes préfèrent la dexaméthasone ou la prednisolone, à des doses appropriées. D’autre part, il faut souligner que l’équivalence des différents GCs est basée sur leur action anti-inflammatoire et non sur différents aspects du métabolisme humain. Puisque le cortisol affecte près de 20% du génome humain, diverses réponses de différents GCs sont attendues dans divers tissus. Cette perception est corroborée par le fait que l’administration de dexaméthasone à des patients atteints de CAH a montré une détérioration des indices de résistance à l’insuline par rapport aux autres GCs (44). De plus, l’administration de 2,5 à 7,5 mg de prednisolone, une dose considérée comme normale, exerce un impact négatif de longue date sur le métabolisme osseux (45). Par conséquent, l’hydrocortisone doit être considérée comme la meilleure forme de traitement en cas de traitement par supplémentation en GC. L’avènement de la formule d’hydrocortisone nouvellement synthétisée avec un comprimé par jour et ses premiers résultats positifs chez les patients atteints de CAH sont très prometteurs pour l’avenir (46).
Prise en charge Pendant l’enfance
Les patients atteints de CCNSA chez qui des signes de PP ont été diagnostiqués pendant l’enfance peuvent être traités avec de l’hydrocortisone dans le but de supprimer les hormones surrénales et de prévenir l’avancement rapide de l’âge osseux qui pourrait affecter la taille finale. Chez les patients chez lesquels le traitement à l’hydrocortisone a été initié 1 an avant le début de la puberté et dont l’âge osseux était inférieur à 9 ans, la taille finale est restée dans le potentiel génétique (47). Les études disponibles indiquent que la taille adulte s’approchait de la taille cible attendue chez les patients qui étaient étroitement surveillés et qui respectaient strictement les plans de médication (48-50). Une petite étude récente portant sur cinq cas (patients âgés de 6,1 à 9,2 ans) a démontré qu’une dose ultra-faible de dexaméthasone est une option prometteuse pour réduire le stress endogène et ses effets. Il reste à déterminer si cela constitue une approche réaliste et dans quelle mesure la dose administrée doit être administrée (51). Une étude de Nebesio et al. a comparé les effets hormonaux et la pharmacocinétique de l’hydrocortisone, de la prednisolone et de la dexaméthasone chez 9 patients prépubères atteints de CCAH. Ils ont montré que la dexaméthasone était plus puissante que les autres formes pour atteindre des niveaux d’hormones surrénales significativement plus bas, suggérant ainsi que la dexaméthasone est plus efficace pour la suppression de la production d’androgènes surrénaliens (46). Bien entendu, d’autres études sont nécessaires pour vérifier ou rejeter cette conclusion. De plus, dans certains cas, des concentrations élevées d’androgènes peuvent entraîner une stimulation secondaire de l’axe de la GnRH, entraînant une puberté prématurée. Dans un tel cas, un parcours parallèle avec l’analogue de la GnRH peut être prescrit si l’âge osseux est significativement supérieur à l’âge chronologique et / ou si la hauteur finale projetée est disproportionnée par rapport à la hauteur cible.
Une autre considération importante concerne les cas de carence concomitante en GH dans le cas où le traitement par des analogues de la GnRH et la prescription de GH ont abouti à l’atteinte de la hauteur cible (52). Selon les directives actuelles, la thérapie décrite ci-dessus n’est recommandée avec prudence que dans les cas où la hauteur prévue SD est de -2,25 < la hauteur cible ou même inférieure (53). Une autre indication pour commencer le traitement à l’hydrocortisone est une réponse inadéquate au cortisol après la stimulation par l’ACTH (36).
Le traitement GC préféré chez les enfants est généralement l’hydrocortisone à 10-15 mg / m2, divisée en trois doses. Souvent, des doses plus faibles se sont également révélées efficaces, à partir de 6 mg / m2 / jour (34). Le surdosage doit être évité, étant donné qu’il peut entraîner une faible croissance ainsi que des caractéristiques Cushingoïdes. Un modèle de croissance régulier, un âge osseux compatible avec l’âge chronologique et l’absence d’obésité centrale peuvent également servir d’indices cliniques pour une prise en charge appropriée. En ce qui concerne le profil biochimique / hormonal, les niveaux d’androstènedione et de testostérone dans les plages moyennes à supérieures pour l’âge osseux sont considérés comme de meilleurs marqueurs d’un traitement de remplacement adéquat de la GC chez les enfants atteints de NCCAH. Cependant, des niveaux de testostérone supprimés ont été observés chez 10 % des patients atteints du CCNSA, alors qu’un autre 28 % des patients présentaient des concentrations accrues de testostérone, ce phénomène pouvant être attribuable à la variabilité de l’hydrocortisone (54, 55). Les valeurs de DHEAS sont diminuées avec de très petites doses, alors que la suppression des niveaux de 17 OHP et de progestérone nécessite des doses de GC très élevées. Il convient de noter que les valeurs de 17 OHP sont cruciales pour le diagnostic, mais ne sont pas utiles lors du suivi. Il convient de noter que le prélèvement sanguin pour l’évaluation hormonale doit être effectué sans arrêt du traitement. Cependant, en raison de la perplexité de la maladie et de sa nature multiforme, il n’existe pas de lignes directrices spécifiques pour le moment des changements de régime ou l’arrêt du traitement aux glucocorticoïdes chez les enfants.
Prise en charge Pendant l’adolescence
Patientes Traitées Depuis l’enfance
Jusqu’à l’établissement du schéma menstruel normal chez les filles du CCNSA, la poursuite de la SCg qui a commencé pendant l’enfance est fortement recommandée (56). Cependant, les patients adolescents ne montrent souvent pas une observance suffisante de l’administration chronique de médicaments et omettent souvent les doses. De plus, pendant la puberté, la demi-vie de l’hydrocortisone diminue de 50% en raison de l’augmentation des niveaux d’IGF-1, ce qui diminue l’activité de 11ßOHSD, ainsi que de l’augmentation de la clairance du cortisol résultant de l’amplification du taux de filtration glomérulaire (57). Chez la jeune femelle, 2 ans après la ménarche et si des cycles ovulatoires normaux ont été enregistrés, une approche centrée sur le patient des symptômes hyperandrogènes qui peuvent apparaître est fortement recommandée.
S’il y a des résultats hyperandrogènes sévères, tels que l’hirsutisme, l’acné et / ou l’oligoménorrhée, la poursuite du traitement sera envisagée. Il est important de se souvenir de la sensibilité fragile d’un jeune adolescent, qui sera gravement endommagé par les signes « répulsifs” de l’hyperandrogénie. La combinaison de signes hyperandrogènes, de troubles menstruels et d’une mauvaise qualité de vie est bien documentée et elle est particulièrement élevée chez les femmes plus jeunes. Un autre point à considérer est la réponse de la stimulation post-ACTH de la réserve surrénale. Étant donné que le pic de cortisol des femelles pubertaires et adultes après stimulation est inférieur à 496 nmol / L, en cas de stress, un traitement aux stéroïdes doit être administré (58). D’autre part, si aucun des symptômes ci-dessus n’est rencontré chez le jeune patient, le traitement par GC peut être interrompu, après quoi un suivi régulier est conseillé.
Femmes Présentant Une Première Manifestation Pendant l’Adolescence ou Des Symptômes Hyperandrogènes Après l’arrêt du traitement
En présence de symptômes hyperandrogènes, une approche axée sur le patient est fortement recommandée, en se concentrant sur les principales plaintes du patient. C’est bien mieux qu’un modus operandi universel général, car, avec cette dernière approche, le jeune patient sera déçu, ce qui entraînera l’arrêt du traitement et, par conséquent, des résultats néfastes pour la santé à l’âge adulte. Dans les cas d’hirsutisme sévère, une cure de 6 à 12 mois avec des pilules contraceptives orales (OCP) constitue une procédure raisonnable compte tenu des effets bénéfiques des OCP sur la production hépatique de SHBG et de la diminution de la libération d’androgènes par l’ovaire. Une amélioration clinique sera attendue après au moins 2 à 3 mois d’initiation de l’OCPs, tandis que l’utilisation simultanée d’un progestatif aux propriétés androgènes minimales est fortement recommandée. En cas d’échec de l’OCP en première intention, des antiandrogènes (spironolactone, flutamide, bicalutamide, acétate de cyprotérone et finastéride) peuvent être ajoutés au traitement. Selon notre expérience, l’administration de bicalutamide a permis une amélioration significative dans les cas d’acné sévère, mais des résultats similaires n’ont pas été obtenus dans les cas d’hirsutisme sévère. Des approches cosmétiques telles que l’application au laser et les dépilatoires peuvent également être suggérées pour les femmes se plaignant d’une croissance excessive ou indésirable des cheveux ou d’une perte de cheveux du cuir chevelu (alopécie androgénique) (32, 34, 57).
Chez les patients présentant une sécrétion insuffisante de cortisol après stimulation, ou si les OCP et les antiandrogènes ne peuvent pas être tolérés, un traitement par GCs est fortement recommandé (57). Données de New et al. indiquer que les règles irrégulières et l’acné sont inversées dans les 3 mois suivant le début du traitement aux glucocorticoïdes, alors que l’hirsutisme nécessite près de 30 mois (59). Contrairement à l’enfance, à l’adolescence, des stéroïdes à action plus longue sont souvent utilisés et des régimes de 5 mg de prednisolone ou de 0,25 mg de dexaméthasone sont recommandés (53). Cependant, dans le monde réel, les données cliniques ont montré une variété de régimes différents appliqués dans la prise en charge du CCNSA (41).
L’objectif principal du traitement doit être de soulager le patient des symptômes hyperandrogènes. Ainsi, chez les personnes ayant reçu un diagnostic de CCNSA basé uniquement sur des résultats de laboratoire, les caractéristiques cliniques devraient guider les recommandations de prise en charge, car les résultats hormonaux et moléculaires ne prédisent pas nécessairement la gravité clinique. Selon les lignes directrices de l’Endocrine Society, les patients du CCNSA devraient avoir la possibilité d’arrêter le traitement par GC lorsque les symptômes disparaissent (60). Bien entendu, ces patients ne doivent pas être perdus pour le suivi, tandis que le traitement doit être relancé en cas de récidive des symptômes. En outre, en cas d’arrêt du traitement, les patients doivent être informés de la possibilité d’infertilité et doivent être encouragés à consulter un médecin s’ils souhaitent concevoir (19). Il convient de noter que le transfert approprié du patient du pédiatrique à l’endocrinologue adulte doit être effectué, de manière optimale après 1 an de surveillance synchronisée (53, 61).
NCCAH et reproduction
Subfertilité
La subfertilité est couramment rapportée dans les formes classiques de CAH, qui est principalement due à des troubles menstruels, une anovulation chronique et des déformations anatomiques. Le taux de natalité a été estimé à 17% par rapport à 65% de la population témoin (62). Pendant ce temps, les données concernant la fertilité chez les femmes atteintes du CCNSA ont récemment été évaluées en détail et l’incidence estimée de l’infertilité est de 11% chez les femmes du CCNSA, c’est-à-dire relativement plus légère que chez les femmes atteintes du CCNSA, et dans de nombreux cas, elle est facilement résolue. Bidet et coll. évaluation de la fécondité chez 190 femmes atteintes de NCCAH, dont 95 voulaient devenir enceintes. Dans cette population, 187 grossesses chez 85 femmes ont été signalées, ce qui a donné lieu à 141 naissances chez 82 personnes. Il faut souligner que 99 des grossesses (52,9%) ont eu lieu avant le diagnostic de NCCAH, dont trois avec l’application de protocoles d’induction de l’ovulation, le reste étant spontané, tandis que 88 grossesses ont eu lieu après le diagnostic de NCCAH. Dans la grande majorité d’entre eux, la grossesse s’est développée après l’institution d’un traitement à l’hydrocortisone, alors que chez 11 femmes, cela s’est produit spontanément (63, 64). Dans un autre rapport de 22 femmes du CCNSA désirant une grossesse, 12 grossesses ont suivi avec la prednisone (65).
Fausses couches
Dans la cohorte de Bidet et al., le taux de fausse couche était de 6,5 et 26,3% chez les patients traités par GCs et les patients non traités, respectivement (64). L’issue de la grossesse peut même réussir sans traitement aux glucocorticoïdes dans les cas où le CCNSA n’a pas encore été diagnostiqué, comme le rapporte également un rapport de cas de Cuhaci et al. Néanmoins, le même couple a connu deux fausses couches et a signalé une sous-fécondité après cette première grossesse (66). En ce qui concerne la possibilité de grossesses prématurées, telle qu’évaluée par Bidet et al., il ne diffère pas significativement de celui du reste de la population (15).
Planification de la fertilité
Pour les femmes présentant un hyperandrogénie symptomatique ou une infertilité signalée mais qui souhaitent concevoir, un traitement par GC est fortement recommandé. Pendant cette période, les glucocorticoïdes utilisés sont la prednisone ou l’hydrocortisone. L’action la plus importante, car elle guidera les étapes suivantes, est le test génétique des futurs parents. Les femmes atteintes du CCNSA qui désirent concevoir doivent être conscientes du risque de donner naissance à un nourrisson atteint de la forme classique de la maladie. Les résultats des grossesses chez les femmes atteintes du CCNAA, et plus particulièrement l’incidence des nourrissons nés avec le CCNAA, sont estimés dans des études récentes entre 1,5 et 2,5 %, le CCNAA étant d’environ 15 % (36, 64). Bien entendu, cette fréquence dépend de la population de référence, une incidence plus élevée se produisant dans les populations avec des taux de mariages mixtes élevés (36). Les chances prévues que les parents donnent naissance à un enfant atteint de CAH sont de 1 sur 120 pour un génotype paternel inconnu, de zéro si le père n’a pas de mutation pertinente et de 1 sur 4 s’il a une mutation hétérozygote (19). En conséquence, les tests génétiques des partenaires de ces femmes sont essentiels pour évaluer le risque de donner naissance à un enfant avec la forme classique de CAH (64, 67).
Dans le cas où la future mère porte deux mutations NC (tableau 1), il n’y a pas de besoin absolu de tests génétiques chez le futur père, car, s’il est hétérozygote pour les mutations du CYP12A2, la progéniture risque de développer la forme non classique de la maladie à l’avenir. Cependant, il convient de noter que le statut de porteur est de 2 à 15% dans différentes populations et que la moitié de ces individus sont porteurs d’une mutation sévère. De plus, les recommandations définitives concernant les situations où les tests génétiques ne sont pas nécessaires sont difficiles étant donné la corrélation génotype-phénotype imparfaite, en particulier pour les mutations plus légères. Par exemple, la mutation P30L, le plus souvent associée au NCCAH, survient également chez les patients atteints d’HTAC classique. Une grande étude européenne multicentrique a récemment montré que c’était le cas chez 78% des patients. Dans cette étude, la mutation légère V218L a été associée à une mutation classique plutôt qu’à une mutation NCCAH dans 30 % des cas. Par conséquent, tous les patients atteints de NCCAH devraient se voir proposer des conseils génétiques et une évaluation moléculaire des partenaires reproducteurs.
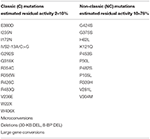
Tableau 1. Mutations du CYP21A2 avec une activité enzymatique résiduelle minimale (C) ou modérée (NC) (14, 68, 69).
En revanche, dans le cas où la future mère est un hétérozygote composé avec une mutation sévère (C) et une mutation NC, le test génétique du futur père est obligatoire. Nous devons garder à l’esprit que les données provenant d’études utilisant des immunoessais ou des procédures LC-MS / MS plus précises mettent constamment en évidence l’incapacité d’exclure de manière fiable l’hétérozygotie en utilisant des valeurs de 17 OHP stimulées par la base ou l’ACTH, en raison du chevauchement significatif avec la plage normale. On peut suggérer l’utilisation d’un test de Synacthène, et s’il n’est pas compatible avec l’hétérozygotie (somme des valeurs de 17 OHP stimulées par la base et le pic < 1,5 nmol/L), les tests ADN pourraient être évités. Cependant, nous devons être très prudents dans l’interprétation de ce test, car il n’a pas été validé dans d’autres études. Par conséquent, l’utilisation du 21-désoxycortisol stimulé par l’ACTH, soit de manière unique, soit dans le cadre d’un rapport stéroïdien ou d’un profil stéroïdien, peut faciliter l’identification biochimique des hétérozygotes à l’avenir, en particulier à mesure que la LC-MS / MS devient plus largement disponible. Si le futur père porte une mutation NC, rien d’autre n’est nécessaire. Inversement, s’il est porteur d’une mutation classique, la question du traitement prénatal du fœtus se posera. Toutes les combinaisons de génotypes possibles et les procédures suggérées sont présentées dans le tableau 2.

Tableau 2. Planification des procédures suggérées pendant la grossesse en fonction du génotype des futurs parents.
Traitement prénatal de la CAH
Le but du traitement prénatal de la CAH est la prévention de la virilisation génitale du fœtus, mais aussi l’atténuation du stress ressenti par les parents susceptibles d’avoir un enfant avec des organes génitaux ambigus (70). La dexaméthasone est utilisée en raison de sa capacité à traverser le placenta et parce qu’elle n’est pas désactivée de la 11β OH steroid déshydrogénase placentaire et ne se lie que très peu à la globuline de liaison au cortisol (CBG) du sang de la mère. La dexaméthasone, en supprimant la sécrétion d’ACTH fœtale, diminue la production d’androgènes fœtaux élevée. Le traitement doit être interrompu dans le cas où l’analyse du caryotype ou de l’ADN révèle un mâle ou une femelle non affectée, respectivement (71). Néanmoins, bien que la virilisation génitale fœtale ait déjà commencé à 6-7 semaines après la conception, les biopsies villeuses choriales pour le diagnostic génétique ne peuvent pas être obtenues avant la 10e−12e semaine. Cet intervalle de temps suggère que toutes les grossesses à risque de CAH virilisante doivent être traitées, même si seulement 1 fœtus sur 8 est affecté et une femme (19, 34, 70).
La question de savoir si cette exposition du fœtus à la dexaméthasone à des fins préventives est médicalement et éthiquement acceptable reste controversée (70). Des études chez l’animal ont démontré que la dexaméthasone au premier trimestre diminue le poids à la naissance, affecte la fonction des cellules bêta pancréatiques rénales et le développement du cerveau, et prédispose à l’anxiété, à l’hypertension et à l’hyperglycémie à l’âge adulte (70). En outre, 10 à 20% des femmes enceintes utilisant un traitement à la dexaméthasone pendant la grossesse se plaignent d’une prise de poids, d’une augmentation de l’appétit, de sautes d’humeur, d’insomnie, d’œdèmes, d’hypertension, d’hyperglycémie et de stries. Cependant, New et al. on a constaté un score de Prader inférieur à la moyenne chez les fœtus traités par la dexaméthasone avant la naissance, mais aucune différence dans les résultats à long terme (72). De plus, dans une revue de Merce Fernandez-Balsells et al., il a été démontré que la dexaméthasone était associée à une réduction de la virilisation fœtale sans effets indésirables maternels ou fœtaux significatifs (63). Cependant, les auteurs de la revue susmentionnée ainsi que des lignes directrices consécutives de l’Endocrine Society soulignent qu’en raison de la petite taille des échantillons dans l’ensemble de la littérature, le sujet reste incertain et des recherches plus approfondies sont clairement nécessaires.
La décision d’initier un traitement ne devrait être prise que dans les grands centres avec une équipe expérimentée et des protocoles approuvés par les commissions d’examen institutionnelles et basés sur les valeurs et les préférences de la famille et avec leur consentement éclairé écrit comme condition préalable (19, 63). Plusieurs spécialistes du domaine indiquent qu’une valeur plus élevée devrait être accordée à la prévention de l’exposition prénatale inutile de la mère et du fœtus à la dexaméthasone plutôt qu’à l’imposition des conséquences émotionnelles des organes génitaux ambigus aux parents et aux patients. Plus important encore, il convient de préciser aux parents que l’administration de dexaméthasone ne modifie pas le statut du patient, mais vise à réduire le besoin de chirurgie plutôt qu’à préserver la vie ou la capacité intellectuelle. Ils doivent également savoir que la probabilité que leur enfant souffre de la forme classique de la maladie est élevée, malgré le traitement. Plus important encore, l’exposition d’un jeune organisme à un GC très puissant pendant une période particulièrement sensible de programmation et de croissance fœtale, qui pourrait bien s’avérer inutile dans le cas d’un fœtus avec le caryotype XY, n’est pas actuellement étayée par des données robustes et incontestables.
Idéalement, il devrait y avoir un diagnostic précoce, effectué via une technique non invasive et avant le début de la virilisation génitale du fœtus. Travaillant dans ce sens, de nouvelles études indiquent l’utilisation de l’ADN fœtal sans cellules obtenu à partir du plasma maternel comme méthode prometteuse qui permettra la détermination du sexe fœtal et le diagnostic de CAH à un âge gestationnel précoce (< 9 semaines). Si cette procédure est largement mise en œuvre dans la pratique clinique, un traitement prénatal inutile à la dexaméthasone sera évité (73).
Traitement Pendant la grossesse
Pendant la grossesse, les femmes atteintes de NCCAH doivent de préférence être traitées avec de l’hydrocortisone, non seulement parce que la grossesse s’accompagne généralement de niveaux de stress élevés, mais aussi à titre préventif contre la forte incidence de fausses couches susmentionnée. Cette forme de glucocorticoïde est favorisée en raison de sa métabolisation par l’enzyme placentaire 11β OH steroid déshydrogénase II, qui empêche les glucocorticoïdes d’avoir un effet quelconque sur le fœtus (71). Au moment de la conception, la dose d’hydrocortisone doit être augmentée à 20-25 mg / jour et des modifications de dose sont effectuées toutes les 6-8 semaines dans le but de maintenir les niveaux de testostérone aux niveaux normaux supérieurs du trimestre de grossesse (74). À titre d’exemple, les valeurs de testostérone et de D4 d’un patient atteint de NCCAH de la conception jusqu’à 3 ans après l’accouchement de deux jumeaux en bonne santé sont présentées (figure 1).
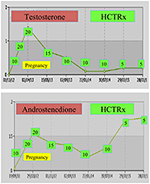
Figure 1. Taux de testostérone et d’andostenedione et dosage ultérieur de l’hydrocortisone (HCTRx), de la conception jusqu’à 3 ans après l’accouchement, administré à un patient atteint de NCCAH.
Autres Problèmes spéciaux
Gestion du stress dans le CCNSA
Lors d’un stress majeur mettant le pronostic vital en jeu, d’une intervention chirurgicale ou d’une maladie grave, les patients atteints du CCNSA traités aux glucocorticoïdes peuvent nécessiter des doses plus importantes ou plus fréquentes de glucocorticoïdes étant donné que leur fonction surrénalienne est iatrogéniquement supprimée. Il est donc crucial d’éduquer les parents de jeunes enfants, ainsi que de rééduquer les patients sur leur transition vers les soins aux adultes, sur le dosage du stress. Pour les patients atteints de NCCAH qui ne sont pas traités par GCs ou en cas d’arrêt du traitement, la réponse du cortisol à l’ACTH doit être évaluée. Près du tiers des patients du CCNSA répondent de manière inadéquate à la stimulation (15, 75). Pour ceux qui répondent normalement à la stimulation par l’ACTH, aucun traitement avec des doses de stress n’est recommandé (19). Un traitement par minéralocorticoïdes n’est nécessaire dans aucun des cas, car ces patients ont une sécrétion normale d’aldostérone et ne développent pas de gaspillage de sel (58). Il convient de noter qu’en cas d’hyperthyroïdie ou d’augmentation du traitement par la lévothyroxine, la clairance du cortisol est augmentée et une crise surrénale peut survenir (58, 76).
Aspects psychobiologiques dans le CCNSA
Meyer-Bahlburg et al. ont dans plusieurs études rapporté un profil psychologique altéré chez les patients atteints de NCCAH en raison d’une carence en 21OH. Plus précisément, les femmes touchées avaient un score de masculinisation / déféminisation plus élevé sur plusieurs mesures du comportement lié au genre par rapport aux femmes témoins normales, bien que nettement moins que chez les femmes ayant une HAC classique. Ils avaient également une orientation bisexuelle ou homosexuelle significativement accrue. Des entretiens cliniques qualitatifs rétrospectifs avec ces femmes ont révélé des antécédents d’inconfort et de stress social liés à leurs expériences de prétraitement avec des signes dépendants des androgènes, tels que l’acné, l’hirsutisme et des difficultés de conception (59, 77, 78). De même, Arlt et coll. a montré que l’état de santé subjectif était significativement altéré dans les huit domaines d’une enquête de santé à court terme, avec les différences les plus importantes, par rapport aux témoins appariés selon l’âge et le sexe, relatives aux domaines de la santé générale, de la vitalité et des limitations de rôle dues à des problèmes émotionnels. Le questionnaire sur l’Échelle d’anxiété et de dépression hospitalière (HAD) a également révélé une augmentation des scores d’anxiété (41). Compte tenu de ces résultats, des paramètres psychologiques pour guider la thérapie devraient être pris en compte chez les femmes atteintes du CCNSA et, dans le contexte de l’approche axée sur le patient, un diagnostic psychologique et un soutien doivent être offerts.
Conclusions
Le NCCAH est considéré comme la forme la plus fréquente et la plus douce de HAC en raison de la rétention d’une activité enzymatique de 20 à 50 %. La plupart des patients peuvent consulter un médecin à n’importe quel stade de leur vie en raison de symptômes liés à un excès d’androgènes. Ceux-ci incluent la puberté prématurée, la croissance rapide, l’hirsutisme, l’acné, les irrégularités menstruelles ou l’infertilité, ainsi que de nombreuses autres manifestations moins importantes. Des valeurs de base tôt le matin de 17 OHP sont recommandées pour un bon test de dépistage initial et une évaluation plus approfondie avec stimulation par l’ACTH et, dans le cas de résultats limites, des tests génétiques. En règle générale, l’androstènedione et non les niveaux de 17 OHP doivent être utilisés pendant le suivi.
La décision de traitement doit être basée sur l’évaluation des faits et doit suivre une approche individualisée. Le traitement n’est pas toujours indiqué sauf si le patient est symptomatique, par exemple, les enfants présentant une apparition précoce et une progression rapide des poils pubiens et corporels, une croissance rapide et / ou un avancement squelettique, ou les femmes présentant une oligoménorrhée, une acné, un hirsutisme, une infertilité ou une combinaison de ces symptômes et d’autres des symptômes susmentionnés. Le conseil génétique est fortement conseillé chez les femmes du CCNSA qui souhaitent concevoir, ainsi que le génotypage du père. La prise en charge globale du patient comprend en outre la prise en charge des complications probables du traitement par glucocorticoïdes ou des manifestations liées au métabolisme de la maladie. Enfin, étant donné que de nombreuses questions thérapeutiques liées à la prise en charge appropriée de ces patients n’ont pas encore été élucidées, il est très important pour le médecin traitant de se tenir au courant de toutes les évolutions dans ce domaine et d’intégrer les nouvelles données dans sa pratique clinique. Certes, d’autres études cliniques dans ce domaine sont essentielles.
Pour résumer, pour la femme du CCNSA, l’approche idéale est une approche sur mesure, intégrant une transition en douceur de sa prise en charge une fois qu’elle est dirigée de l’endocrinologue pédiatrique à l’endocrinologue adulte, ainsi qu’un traitement axé sur les symptômes qui l’accompagnera tout au long de sa vie. Compte tenu des multiples facteurs affectant le système hyperandrogène, il est conseillé d’encourager les patients à adopter un mode de vie plus sain en améliorant leurs habitudes alimentaires, en augmentant l’exercice et en visant la réduction de poids. De plus, dans certains cas, un soutien psychologique est souvent bénéfique. De plus, les spécialistes des domaines impliqués dans le traitement de ce trouble, tels que les dermatologues, les gynécologues et les psychologues, ont besoin d’une compréhension approfondie de la gestion des cas suspects ou déjà diagnostiqués de NCCAH. Pour conclure, gardons à l’esprit la citation perspicace de l’astronome américaine Vera Cooper Rubin: « La science progresse mieux lorsque les observations nous obligent à modifier nos idées préconçues. »
Contributions des auteurs
Tous les auteurs énumérés ont apporté une contribution substantielle, directe et intellectuelle à l’œuvre et l’ont approuvée pour publication.
Déclaration sur les conflits d’intérêts
Les auteurs déclarent que la recherche a été menée en l’absence de relations commerciales ou financières pouvant être interprétées comme un conflit d’intérêts potentiel.
1. Speiser PW, PC blanc. Hyperplasie surrénalienne congénitale. En anglais J Med. (2003) 349:776–88. doi:10.1056/NEJMra021561
Résumé publié/Texte intégral croisé/Google Scholar
2. Turcu AF, Auchus RJ. Adrenal steroidogenesis and congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. (2015) 44:275–96. doi: 10.1016/j.ecl.2015.02.002
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Speiser PW. Nonclassic adrenal hyperplasia. Rev Endocr Metab Disord. (2009) 10:77–82. doi: 10.1007/s11154-008-9097-x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Gidlöf S, Falhammar H, Thilén A, von Döbeln U, Ritzén M, Wedell A, et al. Cent ans d’hyperplasie surrénale congénitale en Suède: une étude de cohorte rétrospective basée sur la population. Diabète Endocrinol de Lancette. (2013) 1:35–42. doi:10.1016/S2213-8587 (13) 70007-X
Résumé publié |Texte intégral croisé |Google Scholar
5. Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, New MI. Haute fréquence de carence en 21-hydroxylase de stéroïdes non classiques. Je suis J Hum Genet. (1985) 37:650–67.
PubMed Abstract | Google Scholar
6. Wilson RC, Nimkarn S, Dumic M, Obeid J, Azar MR, Azar M, et al. Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Mol Genet Metab. (2007) 90:414–21. doi: 10.1016/j.ymgme.2006.12.005
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Miller WL. Clinical review 54: genetics, diagnosis, and management of 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1994) 78:241–6. doi: 10.1210/jcem.78.2.8106606
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Witchel SF, Azziz R. Nonclassic congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2010) 2010:625105. doi: 10.1186/1687-9856-2010-625105
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Wilson RC, Mercado AB, Cheng KC, New MI. Carence en 21-hydroxylase de stéroïdes: le génotype peut ne pas prédire le phénotype. J Clin Endocrinol Metab. (1995) 80:2322–9. doi: 10.1210 / jc.80.8.2322
CrossRef Texte intégral/Google Scholar
10. PC blanc. Dépistage néonatal de l’hyperplasie surrénale congénitale. Nat Rev Endocrinol. (2009) 5:490–8. doi: 10.1038/ nrendo.2009.148
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Kohn B, Levine LS, Pollack MS, Pang S, Lorenzen F, Levy D, et al. Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. (1982) 55:817–27. doi: 10.1210/jcem-55-5-817
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Moran C, Azziz R, Carmina E, Dewailly D, Fruzzetti F, Ibañez L, et al. L’hyperplasie surrénalienne non classique déficiente en 21 hydroxylases est un trouble progressif: une étude multicentrique. Je suis obstétricien Gynécol. (2000) 183:1468–74. doi: 10.1067/ mob.2000.108020
Résumé publié/Texte intégral croisé/Google Scholar
13. Azziz R, Hincapie LA, Knochenhauer ES, Dewailly D, Fox L, Bottes LR. Dépistage de l’hyperplasie surrénale non classique déficiente en 21 hydroxylases chez les femmes hyperandrogènes: une étude prospective. Fertil Steril. (1999) 72:915–25. doi:10.1016/S0015-0282(99) 00383-0
Résumé publié|Texte intégral croisé|Google Scholar
14. Livadas S, Dracopoulou M, Dastamani A, Sertedaki A, Maniati-Christidi M, Magiakou A-M, et al. Le spectre des résultats cliniques, hormonaux et moléculaires chez 280 personnes atteintes d’hyperplasie surrénale congénitale non classique causée par des mutations du gène CYP21A2. Clin Endocrinol (Oxf). (2015) 82:543–9. doi: 10.1111/cen.12543
Résumé publié/Texte intégral croisé/Google Scholar
15. Bidet M, Bellanné-Chantelot C, Galand-Portier M-B, Tardy V, Billaud L, Laborde K, et al. Caractérisation clinique et moléculaire d’une cohorte de 161 femmes non apparentées atteintes d’hyperplasie surrénale congénitale non classique due à un déficit en 21-hydroxylase et de 330 membres de la famille. J Clin Endocrinol Metab. (2009) 94:1570–8. doi: 10.1210 / jc.2008-1582
Résumé publié|Texte intégral croisé/Google Scholar
16. Escobar – Morreale HF, Sanchón R, San Millán JL. Une étude prospective de la prévalence de l’hyperplasie surrénale congénitale non classique chez les femmes présentant des symptômes et des signes hyperandrogènes. J Clin Endocrinol Metab. (2008) 93:527–33. doi: 10.1210 / jc.2007-2053
Résumé publié|Texte intégral croisé/Google Scholar
17. Dewailly D, Vantyghem-Haudiquet MC, Sainsard C, Buvat J, Cappoen JP, Ardaens K, et al. Phénotypes cliniques et biologiques dans le déficit en 21-hydroxylase d’apparition tardive. J Clin Endocrinol Metab. (1986) 63:418–23. doi: 10.1210 / jcem-63-2-418
Résumé publié|Texte intégral croisé|Google Scholar
18. Gönç EN, Ozön ZA, Alikaşifoglu A, Engiz O, Bulum B, Kandemir N. (2011). La progestérone 17-OH sérique basale est-elle un paramètre fiable pour prédire l’hyperplasie surrénale congénitale non classique dans les surrénales prématurées? Turk J Pediatr. 53:274–80.
Résumé publié/Google Scholar
19. Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, et al. Hyperplasie surrénale congénitale due à un déficit en 21-hydroxylase de stéroïdes: une ligne directrice de pratique clinique de la société endocrinienne. J Clin Endocrinol Metab. (2010) 95:4133–60. doi: 10.1210 / jc.2009-2631
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Dacou-Voutetakis C, Dracopoulou M. High incidence of molecular defects of the CYP21 gene in patients with premature adrenarche. J Clin Endocrinol Metab. (1999) 84:1570–4. doi: 10.1210/jc.84.5.1570
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Ambroziak U, Kepczynska-U, kurylović, małunowicz EM, Wójcicka, miskiewicz P, Et al. The diagnosis of nonclassic congénitale hyperplasie du cortex surrénalien due to 21-hydroxylase deficiency, based on sérum basal or post-ACTH stimulation 17-hydroxyprogesterone, can lead to false-positive diagnosis. Clin Endocrinol (Oxf). (2016) 84:23–9. doi: 10.1111 / prix.12935
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Ambroziak U, Kepczynska-U, Kurylowicz, Wysłouch-Cieszyńska A, Małunowicz EM, Bartoszewicz, et al. LC-MS/MS improves screening towards 21-hydroxylase deficiency. Gynecol Endocrinol. (2015) 31:296–300. doi: 10.3109/09513590.2014.994599
PubMed Abstract | CrossRef Full Text | Google Scholar
23. BBC, CC, Goldman MM, Zhang K, Clark-N, Reitz RE Welt CK. Mesure simultanée de treize hormones stéroïdes chez des femmes atteintes du syndrome des ovaires polykystiques et des femmes témoins à l’aide de la chromatographie liquide – spectrométrie de masse en tandem. PLoS Un. (2014) 9: e93805. doi: 10.1371 / journal.pone.0093805
Résumé publié/Texte intégral croisé/Google Scholar
24. Ray JA, Kushnir MM, Yost RA, Rockwood AL, Wayne Meikle A. Amélioration des performances dans la mesure de 5 stéroïdes endogènes par LC-MS / MS combinée à une spectrométrie de mobilité ionique différentielle. Clin Chim Acta. (2015) 438:330–6. doi: 10.1016 / j.cca.2014.07.036
Résumé publié|Texte intégral croisé/Google Scholar
25. Stoupa A, González-Briceño L, Pinto G, Samara-Boustani D, Thalassinos C, Flechtner I, et al. Réponse insuffisante du cortisol au test du tétracosactide (Synacthen®) dans l’hyperplasie surrénale congénitale non classique: une exception à la règle? Horm Res Paediatr. (2015) 83:262–7. doi: 10.1159/000369901
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. (1992) 90:584–95. doi: 10.1172/JCI115897
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Temeck JW, Pang SY, Nelson C, Nouveau MI. Défauts génétiques de la stéroïdogenèse dans la puberté prématurée. J Clin Endocrinol Metab. (1987) 64:609–17. doi: 10.1210 / jcem-64-3-609
Résumé publié|Texte intégral croisé|Google Scholar
28. Voutilainen R, Jääskeläinen J. Surrénarche prématurée: étiologie, résultats cliniques et conséquences. J Biochem stéroïde Mol Biol. (2015) 145:226–36. doi: 10.1016/ j.jsbmb.2014.06.004
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Oberfield SE, Sopher AB, Gerken AT. Approach to the girl with early onset of pubic hair. J Clin Endocrinol Metab. (2011) 96:1610–22. doi: 10.1210/jc.2011-0225
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Janus D, Wojcik M, Malunowicz E, Starzyk JB. Un cas d’adhérences labiales récurrentes chez un enfant de 15 mois présentant une hyperplasie surrénale congénitale non classique asymptomatique due à un déficit en 21-hydroxylase. J Pediatr Endocrinol Metab. (2012) 25:1017–21. doi:10.1515/jpem-2012-0190
Résumé publié/Texte intégral croisé/Google Scholar
31. Gopal-Kothandapani JS, Petkar A, O’Shea E, Banerjee I. Les cheveux périanaux comme une présentation inhabituelle de l’hyperplasie surrénale congénitale non classique. Cas BMJ Rep. (2013) 2013: bcr2013010123. doi: 10.1136/bcr-2013-010123
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Falhammar H, Nordenström A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine. (2015) 50:32–50. doi: 10.1007/s12020-015-0656-0
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Chrousos GP, Loriaux DL, Mann DL, Cutler GB. Late-onset 21-hydroxylase deficiency mimicking idiopathic hirsutism or polycystic ovarian disease. Ann Intern Med. (1982) 96:143–8. doi: 10.7326/0003-4819-96-2-143
CrossRef Full Text | Google Scholar
34. Witchel SF. Non-classic congenital adrenal hyperplasia. Steroids. (2013) 78:747–50. doi: 10.1016/j.steroids.2013.04.010
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Carmina E, Rosato F, Jannì A, Rizzo M, Longo RA. Extensive clinical experience: relative prevalence of different androgen excess disorders in 950 women referred because of clinical hyperandrogenism. J Clin Endocrinol Metab. (2006) 91:2–6. doi: 10.1210/jc.2005-1457
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Moran C, Azziz R, Weintrob N, Witchel SF, Rohmer V, Dewailly D, et al. Résultat reproductif des femmes atteintes d’hyperplasie surrénalienne non classique déficiente en 21-hydroxylase. J Clin Endocrinol Metab. (2006) 91:3451–6. doi: 10.1210 / jc.2006-0062
Résumé publié|Texte intégral croisé/Google Scholar
37. Levin JH, Carmina E, Lobo RA. La sécrétion inappropriée de gonadotrophines chez les patients atteints du syndrome des ovaires polykystiques est-elle similaire à celle des patients atteints d’hyperplasie congénitale des surrénales à l’âge adulte? Fertil Steril. (1991) 56:635–40. doi: 10.1016/S0015-0282(16)54592-0
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Carmina E, Lobo RA. Ovarian suppression reduces clinical and endocrine expression of late-onset congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Fertil Steril. (1994) 62:738–43. doi: 10.1016/S0015-0282(16)56998-2
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Ludwig E. Classification des types d’alopécie androgénétique (calvitie commune) survenant chez le sexe féminin. Br J Dermatol. (1977) 97:247–54. doi: 10.1111/j.1365-2133.1977.tb15179.x
Résumé publié/Texte intégral croisé/Google Scholar
40. Trakakis E, Papadavid E, Dalamaga M, Koumaki D, Stavrianeas N, Rigopoulos D, et al. Prévalence de l’hyperplasie surrénale congénitale non classique due à un déficit en 21-hydroxylase chez les femmes grecques souffrant d’acné: une étude transversale en milieu hospitalier. J Eur Acad Dermatol Vénéréol. (2013) 27:1448–51. doi: 10.1111 / j.1468-3083.2012.04613.x
Résumé publié|Texte intégral croisé|Google Scholar
41. Arlt W, Willis DS, Wild SH, Krone N, Doherty EJ, Hahner S, et al. État de santé des adultes atteints d’hyperplasie surrénale congénitale: une étude de cohorte de 203 patients. J Clin Endocrinol Metab. (2010) 95:5110–21. doi: 10.1210 / jc.2010-0917
Résumé publié|Texte intégral croisé/Google Scholar
42. Saygili F, Oge A, Yilmaz C. Hyperinsulinémie et insensibilité à l’insuline chez les femmes atteintes d’hyperplasie surrénale congénitale non classique due à un déficit en 21-hydroxylase: relation entre les taux sériques de leptine et l’hyperinsulinémie chronique. Horm Res. (2005) 63:270-4. doi: 10.1159/000086363
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Speiser PW, Serrat J, New MI, Gertner JM. Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1992) 75:1421–4. doi: 10.1210/jc.75.6.1421
PubMed Abstract | CrossRef Full Text | Google Scholar
44. Han TS, Stimson RH, Rees DA, Krone N, Willis DS, Conway GS, et al. Régime de traitement aux glucocorticoïdes et résultats pour la santé chez les adultes atteints d’hyperplasie surrénale congénitale. Clin Endocrinol. (2013) 78:197–203. doi: 10.1111/cen.12045
Résumé publié/Texte intégral croisé/Google Scholar
45. Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Utilisation de corticostéroïdes oraux et risque de fractures. J Bone Miner Res. (2000) 15:993-1000. doi: 10.1359 / jbmr.2000.15.6.993
PubMed Abstract | CrossRef Full Text | Google Scholar
46. Nebesio TD, Renbarger JL, Nabhan ZM, Ross SE, Slaven JE, Li L, et al. Differential effects of hydrocortisone, prednisone, and dexamethasone on hormonal and pharmacokinetic profiles: a pilot study in children with congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2016) 2016:17. doi: 10.1186 / l13633-016-0035-5
Résumé publié|Texte intégral croisé|Google Scholar
47. Weintrob N, Dickerman Z, Sprecher E, Galatzer A, Pertzelan A. Déficit non classique en 21-hydroxylase dans la petite enfance et l’enfance: l’effet du temps d’initiation du traitement sur la puberté et la hauteur finale. Eur J Endocrinol. (1997) 136:188–95. doi: 10.1530/eje.0.1360188
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Bonfig W, Bechtold S, Schmidt H, Knorr D, Schwarz HP. Reduced final height outcome in congenital adrenal hyperplasia under prednisone treatment: deceleration of growth velocity during puberty. J Clin Endocrinol Metab. (2007) 92:1635–9. doi: 10.1210/jc.2006-2109
PubMed Abstract | CrossRef Full Text | Google Scholar
49. Dörr HG. Croissance chez les patients présentant une hyperplasie surrénale congénitale classique due à un déficit en 21-hydroxylase. Horm Res. (2007) 68 (Suppl 5): 93-9. doi:10.1159/000110587
Résumé publié / Texte intégral croisé /Google Scholar
50. Hoepffner W, Kaufhold A, Willgerodt H, Keller E. Les patients présentant une hyperplasie surrénale congénitale classique due à un déficit en 21-hydroxylase peuvent atteindre leur hauteur cible: l’expérience de Leipzig. Horm Res. (2008) 70:42-50. doi: 10.1159/000129677
PubMed Abstract | CrossRef Full Text | Google Scholar
51. van der Kaay D, van den Akker E. Ultralow-dose dexamethasone to preserve endogenous cortisol stress response in nonclassical congenital adrenal hyperplasia: a new promising treatment. Int J Endocrinol Metab. (2014) 12:e14657. doi: 10.5812/ijem.14657
CrossRef Full Text | Google Scholar
52. Lin-Su K, Vogiatzi MG, Marshall I, Harbison MD, Macapagal MC, Betensky B, et al. Treatment with growth hormone and luteinizing hormone releasing hormone analog improves final adult height in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. (2005) 90:3318–25. doi: 10.1210/jc.2004-2128
PubMed Abstract | CrossRef Full Text | Google Scholar
53. McCann-Crosby B, Chen M-J, Lyons SK, Lin Y, Axelrad M, Dietrich JE, et al. Nonclassical congenital adrenal hyperplasia: cibles de traitement et de transition. Pediatr Endocrinol Rev. (2014) 12:224-38.
Résumé publié/Google Scholar
54. Ogilvie CM, Crouch NS, Rumsby G, Creighton SM, Liao L-M, Conway GS. Hyperplasie surrénale congénitale chez l’adulte: examen des problèmes médicaux, chirurgicaux et psychologiques. Clin Endocrinol. (2006) 64:2–11. doi: 10.1111/j.1365-2265.2005.02410.x
PubMed Abstract | CrossRef Full Text | Google Scholar
55. Mah PM, Jenkins RC, Rostami-Hodjegan A, Newell-Price J, Doane A, Ibbotson V, et al. Weight-related dosing, timing and monitoring hydrocortisone replacement therapy in patients with adrenal insufficiency. Clin Endocrinol. (2004) 61:367–75. doi: 10.1111/j.1365-2265.2004.02106.x
PubMed Abstract | CrossRef Full Text | Google Scholar
56. Diaz A, Laufer M. Culasse LLAA de PC sur A, Soins AC de O et GC sur AH. Menstruation chez les filles et les adolescentes: utilisation du cycle menstruel comme signe vital. Pédiatrie. (2006) 118:2245–50. doi: 10.1542/ peds.2006-2481
CrossRef Texte intégral/Google Scholar
57. Merke DP, Poppas DP. Prise en charge des adolescents atteints d’hyperplasie surrénale congénitale. diabète Endocrinol de lancette. (2013) 1:341–52. doi: 10.1016/S2213-8587(13)70138-4
PubMed Abstract | CrossRef Full Text | Google Scholar
58. Verma S, Green-Golan L, VanRyzin C, Drinkard B, Mehta SP, Weise M, et al. Adrenomedullary function in patients with nonclassic congenital adrenal hyperplasia. Horm Metab Res. (2010) 42:607–12. doi: 10.1055/s-0030-1253385
PubMed Abstract | CrossRef Full Text | Google Scholar
59. New MI. Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2006) 91:4205–14. doi: 10.1210/jc.2006-1645
PubMed Abstract | CrossRef Full Text | Google Scholar
60. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2018) 103:4043–88. doi: 10.1210/jc.2018-01865
PubMed Abstract | CrossRef Full Text | Google Scholar
61. Kruse B, Riepe FG, Krone N, Bosinski HAG, Kloehn S, Partsch CJ, et al. Congenital adrenal hyperplasia – how to improve the transition from adolescence to adult life. Exp Clin Endocrinol Diabetes. (2004) 112:343–55. doi: 10.1055/s-2004-821013
PubMed Abstract | CrossRef Full Text | Google Scholar
62. Gastaud F, Bouvattier C, Duranteau L, Brauner R, Thibaud E, Kutten F, et al. Résultats sexuels et reproductifs altérés chez les femmes présentant des formes classiques d’hyperplasie surrénalienne congénitale. J Clin Endocrinol Metab. (2007) 92:1391–6. doi: 10.1210 / jc.2006-1757
Résumé publié|Texte intégral croisé/Google Scholar
63. Mercè Fernández-Balsells M, Muthusamy K, Smushkin G, Lampropulos JF, Elamin MB, Abu Elnour NO, et al. Utilisation prénatale de dexaméthasone pour la prévention de la virilisation dans les grossesses à risque d’hyperplasie surrénale congénitale classique en raison d’un déficit en 21-hydroxylase (CYP21A2): une revue systématique et des méta-analyses. Clin Endocrinol. (2010) 73:436–44. doi: 10.1111 /j.1365-2265.2010.03826.x
CrossRef Texte intégral|Google Scholar
64. Bidet M, Bellanné-Chantelot C, Galand-Portier MB, Golmard JL, Tardy V, Morel Y, et al. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2010) 95:1182–90. doi: 10.1210/jc.2009-1383
PubMed Abstract | CrossRef Full Text | Google Scholar
65. Birnbaum MD, Rose LI. Late onset adrenocortical hydroxylase deficiencies associated with menstrual dysfunction. Obstet Gynecol. (1984) 63:445–51.
PubMed Abstract | Google Scholar
66. Cuhaci N, Aydin C, Yesilyurt A, Pinarli FA, Ersoy R, Cakir B. Nonclassical congenital adrenal hyperplasia and pregnancy. Case Rep Endocrinol. (2015) 2015:296924. doi: 10.1155/2015/296924
PubMed Abstract | CrossRef Full Text | Google Scholar
67. Deneux C, Tardy V, Anne D, Mornet E, Billaud L, Charron D, et al. Corrélation phénotype-génotype chez 56 femmes atteintes d’hyperplasie surrénale congénitale non classique due à un déficit en 21-hydroxylase. J Clin Endocrinol Metab. (2001) 86:207–13. doi: 10.1210/ jcem.86.1.7131
Résumé publié|Texte intégral croisé/Google Scholar
68. Simonetti L, Bruque CD, Fernández CS, Benavides-Mori B, Delea M, Kolomenski JE, et al. Mise à jour sur la mutation du CYP21A2: Analyse complète des bases de données et des variantes génétiques publiées. Hum Mutat. (2018) 39:5–22. doi: 10.1002/ humu.23351
PubMed Abstract | CrossRef Full Text | Google Scholar
69. OMIM Entry – * 613815 – CYTOCHROME P450 FAMILY 21 SUBFAMILY A POLYPEPTIDE 2; CYP21A2.
Google Scholar
70. Miller WL, Witchel SF. Prenatal treatment of congenital adrenal hyperplasia: risks outweigh benefits. Am J Obstet Gynecol. (2013) 208:354–9. doi: 10.1016/j.ajog.2012.10.885
PubMed Abstract | CrossRef Full Text | Google Scholar
71. Lekarev O, New MI. Adrenal disease in pregnancy. Best Pract Res Clin Endocrinol Metab. (2011) 25:959–73. doi: 10.1016/j.beem.2011.08.004
PubMed Abstract | CrossRef Full Text | Google Scholar
72. New MI, Abraham M, Yuen T, Lekarev O. An update on prenatal diagnosis and treatment of congenital adrenal hyperplasia. Semin Reprod Med. (2012) 30:396–9. doi: 10.1055/s-0032-1324723
PubMed Abstract | CrossRef Full Text | Google Scholar
73. Kazmi D, Bailey J, Yau M, Abu-Amer W, Kumar A, Low M, et al. New developments in prenatal diagnosis of congenital adrenal hyperplasia. J Steroid Biochem Mol Biol. (2017) 165:121–3. doi: 10.1016/j.jsbmb.2016.06.016
PubMed Abstract | CrossRef Full Text | Google Scholar
74. Clayton PE, Miller WL, Oberfield SE, Ritzén EM, Sippell WG, Speiser PW, et al. Déclaration de consensus sur le déficit en 21-hydroxylase de la Société Européenne d’Endocrinologie Pédiatrique et de la lawson wilkins pediatric endocrine society. Horm Res. (2002) 58:188-95. doi:10.1159/000065490
Résumé publié / Texte intégral croisé /Google Scholar
75. Nandagopal R, Sinaii N, Avila NA, Van Ryzin C, Chen W, Finkielstain GP, et al. Phenotypic profiling of parents with cryptic nonclassic congénital surrenal hyperplasia: findings in 145 unrelated families. Eur J Endocrinol. (2011) 164:977–84. doi:10.1530/EJE-11-0019
Résumé publié /Texte intégral croisé /Google Scholar
76. Takasu N, Nakachi K, Higa H. Le développement de l’hyperthyroïdie de Graves a provoqué une crise surrénale chez un patient présentant un déficit non classique en 21-hydroxylase non reconnu auparavant. Stagiaire Med. (2010) 49:1395–400. doi: 10.2169 / médecine interne.49.3573
PubMed Abstract | CrossRef Full Text | Google Scholar
77. Meyer-Bahlburg HFL, Dolezal C, Baker SW, Carlson AD, Obeid JS, New MI. Prenatal androgenization affects gender-related behavior but not gender identity in 5-12-year-old girls with congenital adrenal hyperplasia. Arch Sex Behav. (2004) 33:97–104. doi: 10.1023/B:ASEB.0000014324.25718.51
CrossRef Full Text | Google Scholar
78. Meyer-Bahlburg HFL, Dolezal C, Baker SW, Nouveau MI. Orientation sexuelle chez les femmes atteintes d’hyperplasie surrénale congénitale classique ou non classique en fonction du degré d’excès d’androgènes prénataux. Le Sexe de l’arche Se comporte. (2008) 37:85–99. doi: 10.1007/s10508-007-9265-1
Résumé PubMed |Texte intégral CrossRef|Google Scholar