La estructura primaria de una proteína se define como la secuencia de aminoácidos de los que está compuesta. Esta secuencia determina en última instancia la forma que adopta la proteína, de acuerdo con las limitaciones espaciales en la disposición de los átomos en la proteína, las propiedades químicas de los residuos de aminoácidos componentes y el entorno de la proteína.
Los enlaces peptídicos que unen los residuos de aminoácidos en un polipéptido se forman en una reacción de condensación entre el grupo carboxilo ácido de un aminoácido y el grupo amino básico de otro aminoácido. En el contexto de un péptido, el grupo de amidas (CO–NH) se conoce como el grupo de péptidos.
Crucial para comprender la estructura de la proteína es conocer la estructura del enlace peptídico. Linus Pauling, en la década de 1930, utilizó la difracción de rayos X para examinar la naturaleza del enlace peptídico formado entre dos aminoácidos. Informó que el grupo peptídico (CO–NH) tiene una estructura plana rígida. Esta estructura se debe a las interacciones entre los electrones del doble enlace del grupo carbonilo y los del enlace C–N (Figura 2), de modo que este último adquiere propiedades parciales de doble enlace (alrededor del 40%).

Este efecto es un ejemplo de resonancia que puede considerarse como un intercambio de electrones entre enlaces. Dado que los enlaces simples entre dos átomos son más largos que los enlaces dobles entre los mismos dos átomos, las longitudes de los enlaces C–N y C=O en el grupo peptídico difieren de las observadas para estos enlaces en otros contextos donde no se produce resonancia. Por lo tanto, el carácter de doble enlace parcial de C–N en el grupo peptídico significa que este enlace es más corto de lo que se predeciría para un enlace simple C–N, mientras que el enlace C=O, que tiene un carácter de enlace simple parcial debido a la resonancia, es más largo de lo que se predeciría para un enlace doble C=O. Las longitudes de enlace en el grupo de péptidos se indican en la Figura 3. Compare el enlace C–N del grupo peptídico con el enlace entre N y Ca (el átomo C al que están unidos el grupo amino y el grupo carboxilo).

Hay dos conformaciones posibles del enlace peptídico plano: en el grupo de péptidos trans, los átomos de Ca están en lados opuestos del enlace peptídico (Figura 3a) y en el grupo de péptidos cis, los átomos de Ca están en el mismo lado del enlace peptídico (Figura 3b).
-
Teniendo en cuenta la disposición espacial y la proximidad de los átomos en las conformaciones cis y trans del enlace peptídico, ¿qué conformación cree que se favorecería?
-
La conformación trans sería energéticamente más favorable que la conformación cis, ya que minimiza los obstáculos estéricos.
en General, los enlaces peptídicos en la conformación trans. Sin embargo, las formas cis pueden ocurrir en enlaces peptídicos que preceden a un residuo de prolina. En tales casos, la forma cis es más estable de lo habitual, ya que la cadena lateral prolina ofrece menos obstáculos. Sin embargo, los enlaces peptídicos cis ocurren solo en aproximadamente el 10% de los casos de enlaces peptídicos que preceden a los residuos de prolina.
Teniendo en cuenta la naturaleza plana del grupo peptídico, se puede ver que una cadena polipeptídica tiene una columna vertebral que consiste en una serie de grupos peptídicos planos rígidos unidos por los átomos de Ca. La Figura 4 muestra parte de un polipéptido con dos grupos peptídicos planos en la conformación trans. Tenga en cuenta que, aunque no se permite la rotación sobre los enlaces peptídicos, existe un potencial de rotación alrededor de los enlaces Ca–N y Ca–C. Los ángulos de rotación , denominados ángulos de torsión, sobre estos enlaces especifican la conformación de una columna vertebral polipeptídica. Los ángulos de torsión de los enlaces Ca–N y Ca–C se denominan ɸ (phi) y ψ. (psi), respectivamente, y se definen como 180° cuando el polipéptido está en la conformación plana extendida, como se ilustra en la Figura 4.
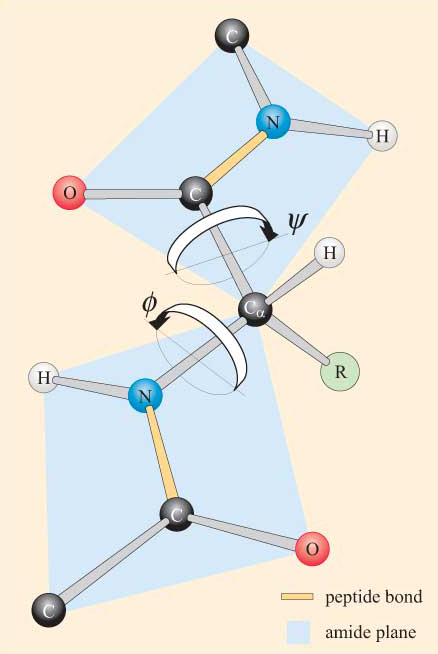
No te sorprenderá saber que las restricciones estéricas se aplican a ɸ y ψ.
Como resultado de estas restricciones estéricas, solo se permiten ciertos valores de ψ y ψ, y por lo tanto conformaciones del péptido, mientras que otros no.
Es posible calcular estos valores permitidos para un residuo dado en el contexto de un polipéptido. Este cálculo se realiza determinando primero las distancias entre todos los átomos no enlazantes en dos grupos de péptidos vecinos (como los de la Figura 4) en todos los valores posibles de ψ y ψ. Se hace más fácilmente para un polipéptido que contiene solo un tipo de aminoácido. Una gráfica conformacional de ψ contra ψ para un residuo en particular se conoce como gráfica de Ramachandran (por su inventor, G. N. Ramachandran). Esta gráfica nos permite identificar aquellas conformaciones (es decir, para un valor particular de ψ y ψ) que son estéricamente favorables o desfavorables (como en la Figura 5), de acuerdo con los siguientes criterios:
-
Cuando no hay conflicto entre los radios de van der Waals de átomos no enlazantes, se ‘permite’una conformación. Estas conformaciones se encuentran en las áreas azules de la Figura 5.
-
Las conformaciones que requieren distancias interatómicas en el límite de lo que es permisible se definen como conformaciones de «límite exterior». Se encuentran en las áreas verdes de la Figura 5.
-
Las conformaciones teóricas que requieren que dos átomos que no se unen estén más cerca entre sí de lo que permiten sus radios de van der Waals están «prohibidas» de forma sterica. Estos se encuentran en las áreas blancas de la Figura 5.
Observe que los valores de ɸ y ψ en la Figura 5 varían de −180º a +180º. Girar el grupo de péptidos a través de 360º, por supuesto, lo devolverá a su posición inicial, y −180º y +180º corresponden a la misma posición. Por lo tanto, la franja verde en la esquina inferior izquierda de la gráfica en la Figura 5 es contigua con el campo en la esquina superior izquierda.

-
Utilice la Figura 5 para determinar si los siguientes valores de ɸ y ψ son estéricamente favorable o desfavorable: (a) ɸ = 90º y ψ = 90º; (b) ɸ = −90º y ψ = 90º.
-
(a) Desfavorable; (b) favorable.
Se pueden construir parcelas de Ramachandran para polímeros de cada uno de los 20 aminoácidos. Es significativo observar que las parcelas de Ramachandran para muchos residuos de aminoácidos son generalmente muy similares, teniendo solo tres regiones con conformaciones favorables o toleradas (marcadas 1-3 en la parcela de poli-l-alanina en la Figura 5). Sin embargo, existen diferencias. Por ejemplo, cuando la cadena lateral (R en la Figura 4) está ramificada cerca de Ca, como en el caso de la treonina, ocupa más espacio cerca de la columna vertebral del péptido y restringe el acercamiento de los átomos en los grupos de péptidos vecinos. Como resultado, las conformaciones permitidas (ángulos XT y ψ) son más restringidas para polipéptidos de aminoácidos ramificados.
-
La prolina también es bastante diferente de otros aminoácidos en términos de conformaciones permitidas y solo para poliprolina, se toleran valores de −85º a −35º. Pensando en la estructura de prolina, ¿cómo puede explicar este rango relativamente estrecho de valores permitted permitidos?
-
La cadena lateral de prolina está unida covalentemente al N del grupo amino, por lo que en la poliprolina, habrá menos libertad de rotación sobre el enlace Ca–N que con otros aminoácidos. En consecuencia, los valores de ɸ permitidos serán relativamente limitados en comparación con otros aminoácidos.
-
La figura 6 muestra la gráfica de Ramachandran para residuos de glicina en una cadena de polipéptidos. Las regiones están codificadas por colores como en la figura 5. ¿Qué puedes decir sobre las conformaciones que adopta la glicina? Considere la estructura de la glicina. ¿Por qué la glicina difiere de los otros residuos con respecto a sus conformaciones?
-
La glicina tiene una libertad conformacional mucho mayor que otros residuos de aminoácidos, porque está menos obstaculizada stericalmente.

Las gráficas de Ramachandran en las Figuras 5 y 6 se han generado para, respectivamente, l-alanina y l-glicina sobre la base de las distancias límite permitidas y externas para los contactos interatómicos, determinadas a partir de valores conocidos para los radios de van der Waals de los átomos (Tabla 1).
Tabla 1 Distancias de Van der Waals para contactos interatómicos.
| Contacto | Normalmente permitido / Å | límite Exterior / Å | |
|---|---|---|---|
| H···H | 2.0 | 1.9 |
3.0 |
Por lo tanto, son gráficos conformacionales predictivos en lugar de reales. Podemos, por supuesto, usar difracción de rayos X para determinar experimentalmente los valores ‘reales’ de ψ y ψ para residuos en un polipéptido. En la Figura 7, los valores ers y ψ para todos los residuos (con la excepción de glicina y prolina) en varias estructuras diferentes se han determinado mediante difracción de rayos X de alta resolución y se han trazado en una gráfica de Ramachandran. Podemos ver que hay una correspondencia sorprendente entre las conformaciones predichas y las reales. Observe, sin embargo, que hay algunos residuos cuyas conformaciones se asignan a las áreas «prohibidas». La mayoría de estos residuos se mapean en la región entre las regiones «permitidas» 2 y 3, alrededor de ψ = 0.

-
Mire de nuevo la figura 4 e imagine que puede girar el grupo de péptidos superior a 180° para que ψ = 0. ¿Qué grupos pueden entrar en conflicto en esta conformación?
-
Los grupos N-H de grupos de péptidos adyacentes entrarán en conflicto entre sí, siendo forzados a estar muy cerca.
El conflicto asociado con estas conformaciones se puede acomodar mediante un pequeño grado de torsión del enlace peptídico. Por lo tanto, en tales conformaciones, el grupo peptídico se tuerce fuera de su conformación plana habitual.
Un número limitado de conformaciones «prohibidas» de residuos particulares puede tolerarse en un polipéptido si la conformación adoptada, en su conjunto, es energéticamente favorable. Un polipéptido tenderá a plegarse de tal manera que adopte la conformación más estable. En esta conformación, el polipéptido minimiza su energía libre. En las siguientes secciones, veremos este nivel más alto de estructura proteica.