- Definiciones y Prevalencia
- Diagnóstico
- Manifestaciones de NCCAH en mujeres
- Manifestaciones en la infancia
- Manifestaciones en la Adolescencia y la Vida Adulta
- Manejo de la AHNC Desde la Primera Manifestación hasta la Vida Adulta
- Manejo Durante la infancia
- Manejo Durante la adolescencia
- Pacientes tratados Desde la infancia
- Mujeres Con Primera Manifestación Durante la Adolescencia o Síntomas Hiperandrogénicos Después de la Interrupción del Tratamiento
- NCCAH y reproducción
- Subfertilidad
- Abortos espontáneos
- Planificación de la fertilidad
- Tratamiento prenatal de la HSC
- Tratamiento Durante el embarazo
- Otros problemas especiales
- Control del estrés en NCCAH
- Aspectos psicobiológicos en NCCAH
- Conclusiones
- Contribuciones de los autores
- Declaración de Conflicto de Intereses
Definiciones y Prevalencia
La hiperplasia suprarrenal congénita (CAH) abarca una familia de trastornos autosómicos recesivos caracterizados por una síntesis de cortisol de leve a aguda debido a una deficiencia en una de las cinco enzimas esteroidogénicas suprarrenales necesarias para la producción de cortisol (1, 2). Convencionalmente, la CAH se divide en (a) clásica (CCAH), que se presenta con pérdida de sal o la forma virilizante simple que se manifiesta al nacer y/o en el período neonatal, y (b) no clásica (NCCAH), que representa una forma menos grave del trastorno que carece de ambigüedad genital, no es inmediatamente peligrosa para la vida y se presenta más adelante en la vida, o permanece asintomática, o se diagnostica erróneamente como una enfermedad diferente (3). Sin embargo, los límites entre las diferentes formas de HSC no están estrictamente definidos, lo que tiende a aumentar los desafíos asociados con este trastorno. Por lo tanto, es aconsejable considerar la HSC como un continuo de fenotipos, de graves a leves o asintomáticos (3).
La forma más común de CAH es causada por la deficiencia de 21-hidroxilasa (21OHD), lo que resulta en una conversión deficiente o nula de 17 hidroxiprogesterona (17 OHP) a 11 desoxicortisol y de progesterona a desoxicorticosterona. El bloqueo de la conversión de esteroides resulta en una mayor producción de precursores de andrógenos, bajo estimulación de CRH-ACTH, lo que conduce a hiperandrogenismo bioquímico, marcado por niveles elevados de 17-OHP. La forma clásica de la enfermedad se presenta en 1 de cada 16.000 nacidos vivos en todo el mundo. Sin embargo, debe mencionarse que la incidencia percibida de la enfermedad está relacionada con el método de cribado utilizado. Por ejemplo, se ha informado de que la incidencia de la forma clásica de HSC en Suecia es de 1:11.500 cuando se utiliza un enfoque de encuesta de casos, que, sin embargo, disminuye a 1:9.800 cuando se aplica el cribado hormonal. Asimismo, la incidencia en Francia y Suiza oscila entre 1:15.472 y 1:23.000 y 1:10.749 y 1:11.661 cuando se utilizan los diferentes métodos de cribado (4).
NCCAH es mucho más frecuente, se presenta en aproximadamente 1 de cada 1.000 caucásicos y con mayor frecuencia en ciertos grupos étnicos, como los judíos asquenazíes (1:27), los hispanos (1: 53), los yugoslavos (1: 62) y los italianos (1:300) (5, 6). Sin embargo, no se dispone de datos sobre la prevalencia de NCCAH basados en diferentes métodos de estimación (encuestas de casos, exámenes hormonales y pruebas moleculares). El NCCAH se considera el trastorno endocrino autosómico recesivo más común con una frecuencia portadora de 1:25 a 1: 10. En NCCAH debido a 21-OHD, se estima que la actividad enzimática residual es de alrededor del 10-70% (7-9).
Diagnóstico
En comparación con el diagnóstico de la forma clásica de la enfermedad, que se realiza al nacer o durante el período neonatal debido a la ambigüedad genital y / o los síntomas de pérdida de sal o a través de programas de detección empleados en algunos países, la mayoría de los casos de AHNCC no son fácilmente detectables (4, 10). Además, muchos individuos permanecen asintomáticos durante la infancia y la adolescencia, tienen una función reproductiva normal y solo se dan cuenta de la NCCAH debido al diagnóstico de otro miembro de la familia y las pruebas consiguientes (11). Sin embargo, la mayoría de las mujeres con NCCAH buscan asistencia médica cuando experimentan síntomas de exceso de andrógenos y, cuando la sospecha clínica incita a las pruebas, es más probable que los niveles basales elevados de 17 OHP apunten a un diagnóstico de NCCAH.
De hecho, las guías clínicas propuestas por la Endocrine Society recomiendan un valor basal no estimulado de 17 OHP como herramienta de cribado para NCCAH. Los niveles de OHP de la mañana 17 > 6 nmol / L en la fase folicular en mujeres menstruantes deben inducir una evaluación adicional, ya que se ha demostrado que los niveles por encima de 6 nmol/L capturan el 90% de los individuos NCCAH (12, 13). Las mediciones aleatorias de 17 OHP no han demostrado ser útiles, ya que a menudo producen niveles normales en pacientes con NCCAH y, además, son extremadamente altas en la fase lútea en la mitad de las mujeres sanas (13). Sin embargo, en nuestros datos derivados de 280 sujetos con la enfermedad, seis pacientes (2.1%) tenía un valor basal de 17 OHP < 6 nmol / L (14). Además, Bidet et al., en una gran cohorte de mujeres con NCCAH verificado por técnicas moleculares, también se encontraron valores basales de 17 OHP inferiores a 6 nmol/L en el 8% de los sujetos estudiados (15). Por último, en base a los datos recopilados por Speiser et al., el 9% de los individuos con AHNC presentaron valores de 17 OHP inferiores a 2 ng/ml (que corresponde a 6nmol/L). Según otros estudios, un valor basal de 17 OHP entre 5,1 y 9 nmol/L es suficiente para el diagnóstico de AHNCC (13, 16, 17). Recientemente, un nivel de 17 OHP basal de 4.6 nmol/L se sugirió como un umbral para la prueba de ACTH para predecir NCCAH en sujetos con adrenarquia prematura durante la infancia (18).
La suma total de estos hallazgos y sugerencias indica que la selección de pacientes que se someterán a una prueba de estimulación Sinacténica debe evaluarse caso por caso. Para el diagnóstico se requiere un nivel postestimulante de 17 OHP superior a 3 nmol/L (19). Los heterocigotos portadores de una mutación en el CYP21A2 muestran niveles ligeramente elevados de 17 OHP después de la estimulación de ACTH, aunque hay superposición en sujetos no afectados (9). Sin embargo, como Dacou-Voutetakis et al. han señalado, si la suma de los valores basales y post-estimulación de 17 OHP excede 1,5 nmol / L, entonces la posibilidad de heterocigosidad es excepcionalmente alta (20).
Otra consideración importante se refiere a las técnicas utilizadas para la evaluación de 17 OHP. Los niveles de 17 OHP se miden mediante una variedad de métodos de inmunoensayo, pero como se ha demostrado recientemente, los resultados más precisos y confiables se lograron mediante la implementación de la combinación de cromatografía líquida con espectrometría de masas (LC-MS/MS). De hecho, se encontraron muchas mediciones de falsos positivos de 17 OHP cuando se compararon las mediciones de LC-MS/Ms con los métodos estándar (21) Sin embargo, estos últimos procedimientos no se utilizan universalmente, en caso de un diagnóstico incierto, proporcionarán resultados más precisos. Cabe destacar que aún no se han definido los umbrales de detección y diagnóstico para LC-MS/MS cuantificados de 17 OHP. Es probable que estos umbrales sean más bajos que los establecidos con los inmunoensayos debido a la especificidad mejorada de LC-MS/MS, un método menos propenso a la reactividad cruzada y a las interferencias (22, 23). Queda por dilucidar si se requiere un perfil de esteroides urinarios para el diagnóstico definitivo (22). En casos límite, es aconsejable obtener un perfil adrenocortical completo después de la prueba de estimulación de ACTH para diferenciar la deficiencia de 21-hidroxilasa de otros defectos enzimáticos y establecer un diagnóstico firme.
Específicamente, se debe realizar un perfil completo de esteroides en casos equívocos para confirmar la deficiencia de 21-hidroxilasa y excluir otras detectaciones enzimáticas. La inclusión de 17 OHP, cortisol, 11-desoxicorticosterona,11-desoxicortisol,17-OH-pregnenolona, Dehidroepiandrosterona y androstenediona en un perfil de esteroides séricos sería útil para excluir otras causas de HSC, como la deficiencia de 11β-hidroxilasa y, más raramente, la deficiencia de 3β-hidroxisteroide deshidrogenasa o la deficiencia de P450 oxidorreductasa (24). Si bien las pruebas genéticas no se consideran una herramienta de diagnóstico primaria para el NCCAH en este momento, son obligatorias para la confirmación del diagnóstico y para el asesoramiento genético (3).
En lo que respecta a los niveles de cortisol, generalmente se espera un valor post-estimulación de al menos 496 nmol/L. Cabe destacar, según Stoupa et al., el 60% de los 47 niños con NCCAH como resultado de 21 OHD tenían valores bajos de cortisol después de la estimulación, un hallazgo que apunta a la necesidad de una mayor vigilancia para el desarrollo de insuficiencia suprarrenal durante eventos estresantes importantes (25).
Manifestaciones de NCCAH en mujeres
La expresión clínica de NCCAH se caracteriza por un alto nivel de polimorfismo en lo que respecta no solo a la edad de inicio, sino también a los diferentes signos y síntomas. Se reporta que la primera presentación clínica de NCCAH es en el 11% de los casos antes de los 10 años y en el 80% entre los 10 y los 40 años (12). Aún no se ha dilucidado la correlación genotipo-fenotipo en HSC y HCC. Speiser et al. sugieren que la mayor parte de la variabilidad fenotípica en la deficiencia de 21-hidroxilasa, pero no toda, se debe a la variación alélica en el CYP21A2 (26).
Manifestaciones en la infancia
En el período neonatal, por lo general, las mujeres NCCAH permanecen asintomáticas y tienen genitales externos normales. El primer caso de NCCAH reportado es una niña de 6 meses que desarrolló vello púbico (11). Por lo general, los hallazgos clínicos y los síntomas en los casos de AHNC comienzan a partir de los 5 años de edad o incluso más tarde y están relacionados con un aumento de los niveles de andrógenos, aunque también puede ocurrir una deficiencia leve de cortisol en algunos casos (8). En el 92% de los casos de AHNCC, el primer síntoma que se manifiesta es la pubertad prematura (PP), que se presenta en niñas menores de 8 años y en niños menores de 9 años (12) con una prevalencia estimada de entre 10 y 11,3% (15). Cabe destacar que en los niños referidos con PP, según diferentes estudios, la incidencia de NCCAH oscila entre el 5 y el 30% (20, 27). Sin embargo, en base a nuestro análisis de 280 pacientes con confirmación molecular de NCCAH, encontramos que la incidencia de PP en 94 mujeres menores de 8 años fue de hasta el 88%. Por otro lado, un análisis de 45 varones con AHNCN identificó PP en solo el 29% de los sujetos (14). Además, debemos tener en cuenta que aproximadamente la mitad de los sujetos con PP pueden ser portadores heterocigotos de una mutación en el CYP21A2.
Otro aspecto que necesita aclaración es la relación entre PP y pubertad precoz. Se plantea la hipótesis de que, en algunos casos, la aromatización periférica de andrógenos suprarrenales a estrógenos puede activar el eje hipotalámico-pituitario-gonadal, lo que conduce a la pubertad precoz secundaria (28). En tal caso, es necesario un seguimiento periódico. Algunos niños desarrollan hirsutismo, acné y / o crecimiento rápido y manifiestan una estructura alta y un avance en la edad ósea (29). Este último puede resultar en una altura final truncada como consecuencia de una fusión epifisaria rápida. Otros hallazgos clínicos poco frecuentes durante la infancia incluyen adhesión labial, vello perianal, clitoromegalia y ronquera de la voz (30-32).
Manifestaciones en la Adolescencia y la Vida Adulta
Durante la adolescencia y la edad adulta, la HCC en mujeres presenta síntomas hiperandrogénicos que se asemejan al síndrome de ovario poliquístico (SOP) (33). Los síntomas incluyen hirsutismo, acné, alopecia androgénica, anovulación, disfunción menstrual e infertilidad. En un estudio multicéntrico, los síntomas más comunes entre mujeres adolescentes y adultas fueron hirsutismo (59%), oligomenorrea (54%) y acné (33%). Entre 161 mujeres con NCCAH, los síntomas que se presentaron fueron hirsutismo (78%), disfunción menstrual (54,7%) y disminución de la fertilidad (12%) (34). En nuestro grupo de 161 mujeres con la enfermedad, el 63% de la paciente presentaba fenotipo ovárico poliquístico (14). Este hallazgo tiene una implicación viceversa, ya que la incidencia de AHNCN es alta en mujeres con hiperandrogenemia. Ciertamente, el estudio de Witcel et al. reportar una incidencia de NCCAH que varía de 1 a 33% en mujeres hiperandrogénicas es de gran interés. En otro estudio de 950 mujeres que fueron remitidas para la evaluación del exceso de andrógenos, se detectó NCCAH en 4,2% de ellas. Sin embargo, los pacientes de NCCAH eran más jóvenes y más hirsutos en comparación con los otros grupos de pacientes hiperandrogénicos. También se caracterizaron por niveles significativamente más altos de testosterona, testosterona libre y 17 OHP que los pacientes con otros síndromes hiperandrogénicos. En la ecografía ovárica, el 77% de ellos mostró ovarios poliquísticos y el 41% aumentó el tamaño ovárico. En resumen, cumplieron los criterios de SOP según los NIH y Rotterdam con un porcentaje del 56 y el 72,8%, respectivamente (35).
La naturaleza progresiva de la enfermedad se destaca por el hecho de que se ha demostrado que la prevalencia del hirsutismo aumenta con la edad y se ha observado que es rara antes de la pubertad. El hirsutismo es la manifestación clínica más común notificada en pacientes con AHNCC, variando del 71 al 96% (28, 36). Las niñas puberales con AHNCN suelen presentar hirsutismo (37, 38). En el otro extremo del espectro está el tema de la alopecia. Se ha informado de calvicie de patrón masculino en pacientes con NCCAH. La prevalencia de alopecia también parece aumentar con la edad, del 6% en los pacientes durante la segunda década de su vida al 19% en la quinta, lo que indica de nuevo la naturaleza progresiva de la enfermedad (39). El acné se reporta en casi el 33% de los sujetos de NCCAH (36). Sorprendentemente, se detectaron mutaciones del gen CYP21A2 que causan NCCAH en el 4,9% de 123 mujeres adultas que presentaban acné severo (40).
Las irregularidades menstruales que incluyen oligomenorrea o amenorrea secundaria a menudo pueden ser el signo de presentación de NCCAH en individuos post menarquías. Además, el 8-9% de los casos también experimentan amenorrea primaria y, por esta razón, buscan asesoramiento médico por primera vez. Además, entre los individuos de NCCAH, la oligomenorrea es más común que la amenorrea primaria. Por ejemplo, en una serie de Moran et al., entre las mujeres de la NCCAH de 10 a 19 años, la oligomenorrea estaba presente en el 56% de los casos en comparación con solo el 9% de la adolescencia con amenorrea primaria (12). Para concluir, tantos síntomas de NCCAH se parecen a los del SOP que no es sorprendente que NCCAH haya sido apodado el gran imitador del SOP. En cualquier caso, se debe considerar un diagnóstico de NCCAH durante la evaluación de cualquier mujer joven que se derive por síntomas hiperandrogénicos.
En un estudio de Arlt et al., los pacientes con AHNCN se caracterizaron por un IMC significativamente más alto y una estatura final más baja en comparación con los controles emparejados (41). Estos datos, junto con algunos estudios que indican la presencia de insensibilidad a la insulina, sugieren un perfil metabólico desfavorable que podría explicarse fisiopatológicamente por el efecto de concentraciones elevadas de andrógenos y/o como resultado de la terapia con glucocorticoides (42, 43). La osteoporosis también se puede detectar en estos sujetos, probablemente como consecuencia de la terapia con corticosteroides (34).
Manejo de la AHNC Desde la Primera Manifestación hasta la Vida Adulta
Una vez establecido el diagnóstico de AHNC, varios aspectos relacionados con las recomendaciones de tratamiento posteriores merecen ser considerados. La primera pregunta que debe abordarse es si está indicada la terapia con glucocorticoides (GC). El razonamiento general detrás de este enfoque es que al proporcionar concentraciones suficientes de cortisol a la paciente, se cubren sus necesidades diarias y, en consecuencia, la estimulación del eje CRH-ACTH se reducirá, lo que disminuirá la producción de andrógenos suprarrenales. La regla general con respecto a este enfoque es que las ECG se administran a dosis de reemplazo y no farmacológicas, mientras que la influencia de la edad, el sexo, los datos de laboratorio, las recomendaciones específicas para el paciente y los objetivos del tratamiento en la terapia de reemplazo con glucocorticoides se tienen seriamente en cuenta. Sin embargo, también debemos ser conscientes de que la terapia de sustitución de CG prolongada puede conducir a hipercortisolemia, con los efectos adversos resultantes bien documentados en todos los aspectos del metabolismo, especialmente el crecimiento, la distribución de grasa y la resistencia a la insulina, así como en el perfil psicológico. Otra desventaja importante de este enfoque es la falta de un índice clínico adecuado o un marcador bioquímico de dosis de reemplazo adecuadas, como los que existen con respecto a los valores de TSH en el hipotiroidismo.
Finalmente, el hecho de que no haya consenso en cuanto a qué CG debe usarse y la ausencia de datos a largo plazo sobre las diferentes modalidades de administración de CG complican aún más las decisiones de los médicos tratantes y la selección de la opción óptima para cada paciente. La hidrocortisona se usa típicamente en niños, ya que se parece más a la hormona natural (cortisol), pero no se considera un enfoque adecuado en adolescentes y mujeres jóvenes debido a la necesidad de dosis diarias múltiples. Por lo tanto, la mayoría de los endocrinólogos adultos prefieren dexametasona o prednisolona, en dosis apropiadas. Por otro lado, hay que señalar que la equivalencia de las diferentes ECG se basa en su acción antiinflamatoria y no en diferentes aspectos del metabolismo humano. Dado que el cortisol afecta a casi el 20% del genoma humano, se esperan respuestas diversas de diferentes GCs en varios tejidos. Corrobora esta percepción el hecho de que la administración de dexametasona a pacientes con HSC ha mostrado deterioro de los índices de resistencia a la insulina en comparación con otros GCs (44). Además, la administración de 2,5–7,5 mg de prednisolona, una dosis considerada normal, ejerce un efecto negativo de larga data sobre el metabolismo óseo (45). Por lo tanto, la hidrocortisona debe considerarse la mejor forma de tratamiento en los casos de terapia de suplementación con CG. El advenimiento de la fórmula de hidrocortisona recién sintetizada con una pastilla al día y sus resultados positivos iniciales en pacientes con HSC son muy prometedores para el futuro (46).
Manejo Durante la infancia
Los pacientes con NCCAH a los que se les diagnostica durante la infancia signos de PP se pueden tratar con hidrocortisona con el objetivo de suprimir las hormonas suprarrenales y prevenir el rápido avance de la edad ósea que podría afectar la estatura final. En aquellos pacientes en los que el tratamiento con hidrocortisona se inició 1 año antes del inicio de la pubertad y que tenían una edad ósea inferior a 9 años, la estatura final permaneció dentro del potencial genético (47). Los estudios disponibles indican que la altura adulta se aproximó a la altura objetivo esperada en pacientes que fueron monitorizados de cerca y que cumplieron estrictamente con los planes de medicación (48-50). Un pequeño estudio reciente de cinco casos (pacientes de 6,1 a 9,2 años de edad) demostró que una dosis ultrabaja de dexametasona es una opción prometedora para reducir el estrés endógeno y sus efectos. Queda por dilucidar si se trata de un enfoque realista y cuánta dosis debe administrarse (51). Un estudio de Nebesio et al. se compararon los efectos hormonales y la farmacocinética de la hidrocortisona, la prednisolona y la dexametasona en 9 pacientes prepúberes con HCC. Mostraron que la dexametasona era más potente que las otras formas para lograr niveles de hormonas suprarrenales significativamente más bajos, lo que sugiere que la dexametasona es más eficaz para la supresión de la producción de andrógenos suprarrenales (46). Por supuesto, se necesitan más estudios para verificar o rechazar este hallazgo. Además, en algunos casos, las concentraciones elevadas de andrógenos pueden conducir a una estimulación secundaria del eje GnRH, lo que conduce a la pubertad prematura. En tal caso, se puede prescribir un curso paralelo con análogo de GnRH si la edad ósea es significativamente mayor que la edad cronológica y / o la altura final proyectada es desproporcionada con la altura objetivo.
Otra consideración importante se refiere a los casos de deficiencia concomitante de GH en el caso de que el tratamiento con análogos de la GnRH y la prescripción de GH condujeran a alcanzar la altura objetivo (52). De acuerdo con las pautas actuales, la terapia descrita anteriormente se recomienda con cautela solo para aquellos casos en los que la altura prevista DE es -2,25 < la altura objetivo o incluso menor (53). Una indicación alternativa para iniciar el tratamiento con hidrocortisona es una respuesta inadecuada de cortisol después de la estimulación de ACTH (36).
El tratamiento con CG preferido en niños suele ser hidrocortisona 10-15 mg / m2, dividida en tres dosis. A menudo, las dosis más bajas también han demostrado ser efectivas, comenzando con 6 mg/m2/día (34). Se debe evitar la sobredosis, ya que puede resultar en un crecimiento deficiente, así como en características cushingoides. Un patrón de crecimiento regular, una edad ósea compatible con la edad cronológica y la ausencia de obesidad central también pueden servir como índices clínicos para un manejo adecuado. Con respecto al perfil bioquímico/ hormonal, los niveles de androstenediona y testosterona en los rangos medio a superior para la edad ósea se consideran mejores marcadores de la terapia de reemplazo de CG adecuada en niños con AHNCN. Sin embargo, se encontraron niveles de testosterona suprimidos en el 10% de los pacientes con AHNC, mientras que otro 28% de los pacientes tenían concentraciones de testosterona aumentadas, este fenómeno posiblemente atribuible a la variabilidad de la hidrocortisona (54, 55). Los valores de DHEAS disminuyen con dosis muy pequeñas, mientras que la supresión de los niveles de 17 OHP y progesterona requiere dosis muy altas de GC. Cabe señalar que los valores de 17 OHP son cruciales para el diagnóstico, pero no son útiles durante el seguimiento. Cabe destacar que la toma de muestras de sangre para la evaluación hormonal debe llevarse a cabo sin interrupción de la terapia. Sin embargo, debido a la perplejidad de la enfermedad y su naturaleza multifacética, no hay pautas específicas para el momento de los cambios de régimen o el cese de la terapia con glucocorticoides en niños.
Manejo Durante la adolescencia
Pacientes tratados Desde la infancia
Hasta el establecimiento del patrón menstrual normal en niñas de NCCAH, se recomienda encarecidamente la continuación de los ECg que comenzaron durante la infancia (56). Sin embargo, los pacientes adolescentes con frecuencia no muestran un cumplimiento suficiente con la administración crónica de medicamentos y a menudo omiten dosis. Además, durante la pubertad, la vida media de la hidrocortisona disminuye en un 50% como resultado del aumento de los niveles de IGF-1, lo que disminuye la actividad de 11ßOHSD, así como debido al aumento del aclaramiento de cortisol derivado de la amplificación de la tasa de filtración glomerular (57). En la mujer joven, 2 años después de la menarquia y si se han registrado ciclos ovulatorios normales, se recomienda encarecidamente un enfoque centrado en el paciente hacia los síntomas hiperandrogénicos que pueden aparecer.
Si hay hallazgos hiperandrogénicos graves, como hirsutismo, acné y/u oligomenorrea, se considerará la continuación del tratamiento. Es importante recordar la sensibilidad frágil de un joven adolescente, que se verá gravemente dañada por los signos «repelentes» del hiperandrogenismo. La combinación de signos hiperandrogénicos, trastornos menstruales y mala calidad de vida está bien documentada y es particularmente alta en mujeres más jóvenes. Otro punto a considerar es la respuesta de la reserva suprarrenal después de la estimulación de ACTH. Dado que el pico de cortisol en mujeres puberales y adultas después de la estimulación es inferior a 496 nmol/L, en casos de estrés, se debe administrar tratamiento con esteroides (58). Por otro lado, si no se presenta ninguno de los síntomas anteriores en el paciente joven, se puede interrumpir el tratamiento con GC, tras lo cual se recomienda un seguimiento regular.
Mujeres Con Primera Manifestación Durante la Adolescencia o Síntomas Hiperandrogénicos Después de la Interrupción del Tratamiento
En presencia de síntomas hiperandrogénicos, se recomienda encarecidamente un enfoque orientado al paciente, centrado en las principales quejas del paciente. Esto es mucho mejor que un modus operandi universal general, ya que, con este último enfoque, el paciente joven se sentirá decepcionado, lo que llevará a la interrupción del tratamiento y, como resultado, a resultados adversos de salud en la edad adulta. En los casos de hirsutismo grave, un curso de 6 a 12 meses con píldoras anticonceptivas orales (PCO) constituye un procedimiento razonable dados los efectos beneficiosos de los PCO en la producción hepática de SHBG y la disminución de la liberación de andrógenos del ovario. Se espera una mejoría clínica después de al menos 2-3 meses de inicio de la terapia con SPCo, mientras que se recomienda encarecidamente el uso simultáneo de un progestágeno con propiedades androgénicas mínimas. Si los PCO fracasan como enfoque de primera línea, se pueden agregar antiandrógenos (espironolactona, flutamida, bicalutamida, acetato de ciproterona y finasterida) al tratamiento. En nuestra experiencia, la administración de bicalutamida ha logrado una mejora significativa en los casos de acné severo, pero no se obtuvieron resultados similares en los casos de hirsutismo severo. También se pueden sugerir enfoques cosméticos como la aplicación de láser y depilatorios para mujeres que se quejan de crecimiento excesivo o no deseado del vello o pérdida de vello en el cuero cabelludo (alopecia androgénica) (32, 34, 57).
En pacientes con secreción inadecuada de cortisol después de la estimulación, o si los OCPs y los antiandrógenos no pueden tolerarse, se recomienda encarecidamente un curso con GCs (57). Datos de New et al. indicar que la menstruación irregular y el acné se revierten dentro de los 3 meses posteriores al inicio de la terapia con glucocorticoides, mientras que el hirsutismo requiere casi 30 meses (59). A diferencia de la infancia, en la adolescencia, a menudo se usan esteroides de acción más prolongada y se recomiendan regímenes de 5 mg de prednisolona o 0,25 mg de dexametasona (53). Sin embargo, en el mundo real, los datos clínicos han mostrado una variedad de regímenes diferentes aplicados en el manejo de NCCAH (41).
El objetivo principal del tratamiento debe ser el alivio del paciente de los síntomas hiperandrogénicos. Por lo tanto, entre los individuos con NCCAH diagnosticado con base únicamente en resultados de laboratorio, las características clínicas deben guiar las recomendaciones de manejo, ya que los hallazgos hormonales y moleculares no necesariamente predicen la gravedad clínica. De acuerdo con las directrices de la Endocrine Society, a los pacientes con HCC se les debe dar la opción de interrumpir el tratamiento con CG cuando los síntomas se resuelvan (60). Por supuesto, estos pacientes no deben perderse en el seguimiento, mientras que el tratamiento debe reiniciarse en caso de recurrencia de los síntomas. Además, en caso de interrupción del tratamiento, se debe informar a las pacientes sobre la posibilidad de infertilidad y se les debe animar a consultar a un médico si desean concebir (19). Cabe destacar que se debe realizar el traslado apropiado del paciente del endocrinólogo pediátrico al endocrinólogo adulto, de manera óptima después de 1 año de monitoreo sincronizado (53, 61).
NCCAH y reproducción
Subfertilidad
La subfertilidad se reporta comúnmente en formas clásicas de HSC, que se debe principalmente a trastornos menstruales, anovulación crónica y deformidades anatómicas. La tasa de natalidad se ha estimado en el 17% en comparación con el 65% de la población de control (62). Mientras tanto, los datos relativos a la fertilidad en mujeres con AHNCN se han evaluado recientemente en detalle y la incidencia estimada de infertilidad es del 11% entre las mujeres con AHNCN, es decir, relativamente más leve que en la HSC, y en muchos casos se resuelve fácilmente. Bidet et al. se evaluó la fertilidad en 190 mujeres que sufrían de NCCAH, 95 de las cuales querían quedar embarazadas. En esta población, se reportaron 187 embarazos en 85 mujeres, que resultaron en 141 nacimientos en 82 individuos. Cabe destacar que 99 de los embarazos (52,9%) ocurrieron antes del diagnóstico de AHNC, tres de ellos con la aplicación de protocolos de inducción de la ovulación, el resto fueron espontáneos, mientras que 88 embarazos tuvieron lugar después del diagnóstico de AHNC. En la gran mayoría de ellas, el embarazo se desarrolló después de la institución de la terapia con hidrocortisona, mientras que en 11 mujeres ocurrió espontáneamente (63, 64). En otro informe de 22 mujeres de NCCAH que deseaban quedar embarazadas, se observaron 12 embarazos con prednisona (65).
Abortos espontáneos
En la cohorte de Bidet et al., la tasa de abortos espontáneos fue de 6,5 y 26,3% en pacientes tratados con ECg y en pacientes no tratados, respectivamente (64). El resultado del embarazo puede incluso ser exitoso sin ningún tratamiento con glucocorticoides en casos en los que el NCCAH aún no se diagnosticó, como también se informó en un informe de caso de Cuhaci et al. Sin embargo, la misma pareja experimentó dos abortos espontáneos y reportó subfertilidad después de este primer embarazo (66). En cuanto a la posibilidad de embarazos prematuros, según lo evaluado por Bidet et al., no difiere significativamente de la del resto de la población (15).
Planificación de la fertilidad
Para aquellas mujeres con hiperandrogenismo sintomático o con infertilidad reportada, pero que desean concebir, la terapia con GC es altamente recomendable. Durante este período, los glucocorticoides utilizados son prednisona o hidrocortisona. La acción más importante, ya que guiará los pasos posteriores, es la prueba genética de los futuros padres. Las mujeres con NCCAH que desean concebir deben ser conscientes del riesgo de dar a luz a un bebé con la forma clásica de la enfermedad. En estudios recientes se estima que el resultado de los embarazos entre las mujeres con AHCC, y más específicamente la incidencia de bebés nacidos con AHCC, está entre el 1,5 y el 2,5%, con un AHCC de aproximadamente el 15% (36, 64). Por supuesto, esta frecuencia depende de la población de referencia, una mayor incidencia ocurre en poblaciones con altas tasas de matrimonios mixtos (36). La probabilidad prevista de que los padres den a luz a un niño con HSC es de 1 en 120 para el genotipo paterno desconocido, cero si el padre no tiene una mutación relevante y 1 en 4 si tiene una mutación heterocigota (19). Como resultado, las pruebas genéticas de las parejas de estas mujeres son esenciales para evaluar el riesgo de dar a luz a un niño con la forma clásica de CAH (64, 67).
En el caso de que la futura madre sea portadora de dos mutaciones NC (Tabla 1), no hay necesidad absoluta de pruebas genéticas en el futuro padre, ya que, si es heterocigoto para las mutaciones del CYP12A2, la descendencia corre el riesgo de desarrollar la forma no clásica de la enfermedad en el futuro. Sin embargo, debe tenerse en cuenta que el estado portador es del 2 al 15% en diferentes poblaciones y la mitad de estos individuos portan una mutación grave. Además, las recomendaciones definitivas con respecto a situaciones en las que no se requieren pruebas genéticas son difíciles dada la correlación imperfecta genotipo-fenotipo, particularmente para mutaciones más leves . Por ejemplo, la mutación P30L, asociada con mayor frecuencia a la HCC, también se presenta en pacientes con HSC clásica . Un gran estudio europeo multicéntrico recientemente demostró que este era el caso en el 78% de los pacientes . En este estudio, la mutación V218L leve se relacionó con la HCC clásica en lugar de la HCC en 30% de los casos. Por lo tanto, a todos los pacientes con NCCAH se les debe ofrecer asesoramiento genético y evaluación molecular de sus parejas reproductoras.
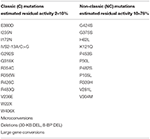
Cuadro 1. Mutaciones del CYP21A2 con actividad enzimática residual mínima (C) o moderada (NC) (14, 68, 69).
Por el contrario, en el caso de que la futura madre sea un heterocigoto compuesto con una mutación grave (C) y una mutación NC, es obligatorio realizar pruebas genéticas al futuro padre. Debemos tener en cuenta que los datos provenientes de estudios que utilizan inmunoensayos o los procedimientos más precisos de LC-MS/MS, destacan consistentemente la incapacidad de excluir de manera confiable la heterocigosidad utilizando valores de 17 OHP estimulados por ACTH o basales, debido a la superposición significativa con el rango normal. Se puede sugerir el uso de una prueba Sinacténica, y si no es compatible con heterocigosidad (suma de valores de 17 OHP estimulados basales y máximos < 1,5 nmol/L), entonces se podría evitar la prueba de ADN. Sin embargo, debemos ser muy cautelosos en la interpretación de esta prueba, ya que no ha sido validada en otros estudios. Por lo tanto, el uso de 21-desoxicortisol estimulado por ACTH, ya sea individualmente o como parte de una relación de esteroides o un perfil de esteroides, puede facilitar la identificación bioquímica de heterocigotos en el futuro, particularmente a medida que LC-MS/MS esté más ampliamente disponible . Si el futuro padre porta una mutación NC, entonces no se necesita nada más. Por el contrario, si es portador de una mutación clásica, surgirá la cuestión relativa al tratamiento prenatal del feto. Todas las combinaciones de genotipos posibles y los procedimientos sugeridos se presentan en la Tabla 2.

la Tabla 2. Planificación de los procedimientos sugeridos durante el embarazo en función del genotipo de los futuros padres.
Tratamiento prenatal de la HSC
El objetivo del tratamiento prenatal de la HSC es prevenir la virilización genital del feto, pero también aliviar el estrés que sienten los padres que probablemente tengan un hijo con genitales ambiguos (70). La dexametasona se usa debido a su capacidad para atravesar la placenta y porque no se desactiva de la deshidrogenasa esteroide 11β OH de la placenta y se une solo mínimamente a la globulina de unión al cortisol (CBG) de la sangre de la madre. La dexametasona, al suprimir la secreción de ACTH fetal, disminuye la producción elevada de andrógenos fetales. El tratamiento debe interrumpirse en caso de que el cariotipo o el análisis de ADN revelen un varón o una mujer no afectada, respectivamente (71). Sin embargo, aunque la virilización genital fetal ya ha comenzado a las 6-7 semanas después de la concepción, las biopsias vellosas coriónicas para el diagnóstico genético no se pueden obtener antes de la semana 10 a 12. Este intervalo de tiempo sugiere que se deben tratar todos los embarazos con riesgo de HSC virilizante, aunque solo 1 de cada 8 fetos esté afectado y mujeres (19, 34, 70).
Sigue siendo controvertido si esta exposición del feto a la dexametasona para medidas preventivas es aceptable desde el punto de vista médico y ético (70). Los estudios en animales han demostrado que la dexametasona en el primer trimestre disminuye el peso al nacer, afecta la función de las células beta pancreáticas renales y el desarrollo cerebral, y predispone a la ansiedad, la hipertensión y la hiperglucemia en la edad adulta (70). Además, el 10-20% de las mujeres embarazadas que usan tratamiento con dexametasona durante el embarazo se quejan de aumento de peso, aumento del apetito, cambios de humor, insomnio, edemas, hipertensión, hiperglucemia y estrías. Sin embargo, New et al. se encontró una puntuación de Prader inferior a la media en fetos tratados con dexametasona antes del parto, pero no hubo diferencia en los resultados a largo plazo (72). Además, en una revisión de Merce Fernandez-Balsells et al., la dexametasona se asoció con una reducción de la virilización del feto sin efectos adversos maternos o fetales significativos (63). Sin embargo, los autores de la revisión mencionada, así como las directrices consecutivas de la Endocrine Society, señalan que, debido a los pequeños tamaños de muestra en todo el cuerpo de la literatura, el tema sigue siendo incierto y es claramente necesario seguir investigando.
La decisión de iniciar el tratamiento debe tomarse solo en centros grandes con un equipo experimentado y protocolos aprobados por Juntas de Revisión Institucionales y basados en los valores y preferencias de la familia y con su consentimiento informado por escrito como requisito previo (19, 63). Varios especialistas en el campo indican que se debe otorgar mayor valor a la prevención de la exposición prenatal innecesaria de la madre y el feto a la dexametasona en lugar de imponer el costo emocional de los genitales ambiguos a los padres y los pacientes. Lo que es más importante, se debe aclarar a los padres que la administración de dexametasona no modifica el estado del paciente, sino que está dirigida a reducir la necesidad de cirugía en lugar de preservar la vida o la capacidad intelectual. También deben saber que la probabilidad de que su hijo sufra la forma clásica de la enfermedad es alta, a pesar del tratamiento. Lo más importante, mientras tanto, la exposición de un organismo joven a un GC muy potente durante un período particularmente sensible de programación y crecimiento fetal, que bien podría resultar inútil en el caso de un feto con el cariotipo XY, no está respaldada por datos sólidos e incuestionables.
Idealmente, debe haber un diagnóstico precoz, esto realizado a través de una técnica no invasiva y antes del inicio de la virilización genital del feto. Trabajando en esta dirección, nuevos estudios apuntan al uso de ADN fetal libre de células obtenido a partir de plasma materno como un método prometedor que permitirá la determinación del género fetal y el diagnóstico de CAH a una edad gestacional temprana (<9 semanas). Si este procedimiento se implementa ampliamente en la práctica clínica, se evitará el tratamiento prenatal innecesario con dexametasona (73).
Tratamiento Durante el embarazo
Durante el embarazo, las mujeres con NCCAH deben ser tratadas preferiblemente con hidrocortisona, no solo porque el embarazo suele ir acompañado de niveles elevados de estrés, sino también como medida preventiva contra la alta incidencia de abortos espontáneos mencionada anteriormente. Esta forma de glucocorticoide es favorecida debido a su metabolización por la enzima placentaria 11β OH esteroide deshidrogenasa II, que evita que el glucocorticoide tenga algún efecto en el feto (71). En el momento de la concepción, la dosis de hidrocortisona debe aumentarse a 20-25 mg/día y las modificaciones de dosis se realizan cada 6-8 semanas con el objetivo de mantener los niveles de testosterona en los niveles normales superiores del trimestre del embarazo (74). A modo de ejemplo, se muestran los valores de testosterona y D4 de un paciente con NCCAH desde la concepción hasta los 3 años posteriores al parto de dos gemelos sanos (Figura 1).
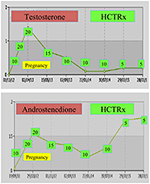
la Figura 1. Niveles de testosterona y andoestendiona y dosificación posterior de hidrocortisona (HCTRx), desde la concepción hasta los 3 años posteriores al parto, administrados a un paciente con NCCAH.
Otros problemas especiales
Control del estrés en NCCAH
Durante el estrés grave que pone en peligro la vida, la cirugía o una enfermedad grave, los pacientes con NCCAH tratados con glucocorticoides pueden requerir dosis más grandes o más frecuentes de glucocorticoides, ya que su función suprarrenal está suprimida iatrogénicamente. Por lo tanto, es crucial educar a los padres de los niños pequeños, así como reeducar a los pacientes en su transición a la atención de adultos, sobre la dosificación del estrés. Se debe evaluar la respuesta del cortisol a la ACTH en los pacientes con AHNC que no reciben tratamiento con GCs o en caso de interrupción del tratamiento. Casi un tercio de los pacientes con AHCC no responden adecuadamente a la estimulación (15, 75). Para aquellos que responden normalmente a la estimulación de ACTH, no se recomienda ningún tratamiento con dosis de estrés (19). No se requiere terapia mineralocorticoide en ninguno de los casos, ya que estos pacientes tienen secreción normal de aldosterona y no desarrollan pérdida de sal (58). Cabe destacar que en el caso del hipertiroidismo o de un aumento en el tratamiento con levotiroxina, el aclaramiento de cortisol aumenta y puede producirse una crisis suprarrenal (58, 76).
Aspectos psicobiológicos en NCCAH
Meyer-Bahlburg et al. en varios estudios, se ha informado de un perfil psicológico deteriorado entre los pacientes con AHNC debido a la deficiencia de 21OH. Más específicamente, las mujeres afectadas tuvieron una puntuación de masculinización / desfeminización más alta en varias medidas de comportamiento relacionado con el género cuando se compararon con las mujeres control normales, aunque marcadamente menos que en las mujeres con HSC clásica. También habían aumentado significativamente la orientación bisexual u homosexual. Las entrevistas clínico-cualitativas retrospectivas con estas mujeres revelaron un historial de incomodidad y estrés social relacionado con sus experiencias previas al tratamiento con signos dependientes de andrógenos, como acné, hirsutismo y dificultades de concepción (59, 77, 78). De manera similar, Arlt et al. mostró que el estado de salud subjetivo se vio afectado significativamente en los ocho dominios de una encuesta de salud a corto plazo, con las diferencias más prominentes, en comparación con los controles emparejados por edad y sexo, en relación con los dominios salud general, vitalidad y limitaciones de roles debido a problemas emocionales. El cuestionario de la Escala de Ansiedad y Depresión Hospitalaria (HADS) también reveló un aumento de las puntuaciones de ansiedad (41). Teniendo en cuenta estos hallazgos, se deben considerar parámetros psicológicos para guiar la terapia en mujeres con AHNCN y, en el contexto del enfoque orientado al paciente, se debe ofrecer un diagnóstico y apoyo psicológicos.
Conclusiones
NCCAH se considera como la forma más frecuente y suave de CAH debido a la retención del 20-50% de actividad enzimática. La mayoría de los pacientes pueden buscar consejo médico en cualquier etapa de su vida debido a síntomas relacionados con el exceso de andrógenos. Estos incluyen pubarquia prematura, crecimiento rápido, hirsutismo, acné, irregularidades menstruales o infertilidad, así como muchas otras manifestaciones menos prominentes. Se recomiendan valores iniciales de 17 OHP a primera hora de la mañana como una buena prueba de detección inicial y una evaluación posterior con estimulación de ACTH y, en el caso de resultados límite, pruebas genéticas. Como regla general, se deben usar niveles de androstenediona y no de 17 OHP durante el seguimiento.
La decisión de tratamiento debe basarse en la evaluación de los hechos y debe seguir un enfoque individualizado. El tratamiento no siempre está indicado a menos que el paciente sea sintomático, por ejemplo, niños con aparición temprana y progresión rápida de vello púbico y corporal, crecimiento rápido y/o avance esquelético, o mujeres con oligomenorrea, acné, hirsutismo, infertilidad o una combinación de estos y otros síntomas mencionados anteriormente. Se recomienda encarecidamente el asesoramiento genético en las mujeres de la NCCAH que desean concebir, así como el genotipado del padre. El tratamiento general del paciente incluye, además, el tratamiento de las complicaciones probables de la terapia con glucocorticoides o de las manifestaciones de la enfermedad relacionadas con el metabolismo. Por último, dado que todavía no se han aclarado muchas cuestiones terapéuticas relacionadas con el manejo adecuado de estos pacientes, es muy importante que el médico tratante se mantenga al día de todos los avances en este campo e integre los nuevos datos en su práctica clínica. Ciertamente, es esencial realizar más estudios clínicos en esta área.
En resumen, para la mujer de NCCAH, el enfoque ideal es uno hecho a medida, que incorpora una transición suave de su manejo una vez que se la remite del endocrinólogo pediátrico al endocrinólogo adulto, junto con un tratamiento orientado a los síntomas que la acompañará a lo largo de su vida. Dados los múltiples factores que afectan el sistema hiperandrogénico, es aconsejable alentar a los pacientes a adoptar un estilo de vida más saludable mejorando sus hábitos alimenticios, aumentando el ejercicio y apuntando a la reducción de peso. Además, en algunos casos, el apoyo psicológico suele ser beneficioso. Además, los especialistas en campos involucrados en el tratamiento de este trastorno, como dermatólogos, ginecólogos y psicólogos, necesitan un conocimiento profundo sobre el manejo de casos sospechosos o ya diagnosticados de NCCAH. Para concluir, tengamos en cuenta la cita perspicaz de la astrónoma estadounidense Vera Cooper Rubin: «La ciencia progresa mejor cuando las observaciones nos obligan a alterar nuestras ideas preconcebidas.»
Contribuciones de los autores
Todos los autores enumerados han hecho una contribución sustancial, directa e intelectual al trabajo y lo han aprobado para su publicación.
Declaración de Conflicto de Intereses
Los autores declaran que la investigación se realizó en ausencia de relaciones comerciales o financieras que pudieran interpretarse como un posible conflicto de intereses.
1. Speiser PW, PC blanco. Hiperplasia suprarrenal congénita. N Engl J Med. (2003) 349:776–88. doi: 10.1056/NEJMra021561
PubMed Abstract | CrossRef Texto Completo | Google Scholar
2. Turcu AF, Auchus RJ. Adrenal steroidogenesis and congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. (2015) 44:275–96. doi: 10.1016/j.ecl.2015.02.002
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Speiser PW. Nonclassic adrenal hyperplasia. Rev Endocr Metab Disord. (2009) 10:77–82. doi: 10.1007/s11154-008-9097-x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Gidlöf S, Falhammar H, Thilén A, von Döbeln U, Ritzén M, Wedell A, et al. One hundred years of congenital suprarrenal hyperplasia in Sweden: a retrospective, population-based cohort study (en inglés). Diabetes Endocrinol Lancet. (2013) 1:35–42. doi: 10.1016/S2213-8587(13)70007-X
PubMed Abstract | CrossRef Texto Completo | Google Scholar
5. Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, New MI. Alta frecuencia de deficiencia no clásica de esteroides 21-hidroxilasa. Soy J Hum Genet. (1985) 37:650–67.
PubMed Abstract | Google Scholar
6. Wilson RC, Nimkarn S, Dumic M, Obeid J, Azar MR, Azar M, et al. Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Mol Genet Metab. (2007) 90:414–21. doi: 10.1016/j.ymgme.2006.12.005
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Miller WL. Clinical review 54: genetics, diagnosis, and management of 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1994) 78:241–6. doi: 10.1210/jcem.78.2.8106606
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Witchel SF, Azziz R. Nonclassic congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2010) 2010:625105. doi: 10.1186/1687-9856-2010-625105
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Wilson RC, Mercado AB, Cheng KC, New MI. Deficiencia de esteroides 21-hidroxilasa: el genotipo puede no predecir el fenotipo. J Clin Endocrinol Metab. (1995) 80:2322–9. doi: 10.1210 / jc.80.8.2322
CrossRef Texto Completo | Google Scholar
10. PC blanco. Detección neonatal de hiperplasia suprarrenal congénita. Nat Rev Endocrinol. (2009) 5:490–8. doi: 10.1038 / nrendo.2009.148
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Kohn B, Levine LS, Pollack MS, Pang S, Lorenzen F, Levy D, et al. Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. (1982) 55:817–27. doi: 10.1210/jcem-55-5-817
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Moran C, Azziz R, Carmina E, Dewailly D, Fruzzetti F, Ibañez L, et al. 21-Hydroxylase-deficient nonclassic adrenal hyperplasia is a progressive disorder: a multicenter study. Am J Obstet Gynecol. (2000) 183:1468–74. doi: 10.1067/mob.2000.108020
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Azziz R, Hincapie LA, Knochenhauer ES, Dewailly D, Fox L, Boots LR. Screening for 21-hydroxylase-deficient nonclassic adrenal hyperplasia among hyperandrogenic women: a prospective study. Fertil Steril. (1999) 72:915–25. doi: 10.1016/S0015-0282(99)00383-0
PubMed Abstract | CrossRef Texto Completo | Google Scholar
14. Livadas S, Dracopoulou M, Dastamani A, Sertedaki A, Maniati-Christidi M, Magiakou A-M, et al. El espectro de hallazgos clínicos, hormonales y moleculares en 280 individuos con hiperplasia suprarrenal congénita no clásica causada por mutaciones del gen CYP21A2. Endocrinol de Clin (Oxf). (2015) 82:543–9. doi: 10.1111 / cen.12543
PubMed Abstract | CrossRef Texto Completo | Google Scholar
15. Bidet M, Bellanné-Chantelot C, Galand-Portier M-B, Tardy V, Billaud L, Laborde K, et al. Caracterización clínica y molecular de una cohorte de 161 mujeres no emparentadas con hiperplasia suprarrenal congénita no clásica debida a deficiencia de 21-hidroxilasa y 330 familiares. J Clin Endocrinol Metab. (2009) 94:1570–8. doi: 10.1210 / jc.2008-1582
PubMed Abstract | CrossRef Texto Completo | Google Scholar
16. Escobar-Morreale HF, Sanchón R, San Millán JL. Estudio prospectivo de la prevalencia de hiperplasia suprarrenal congénita no clásica entre mujeres que presentan síntomas y signos hiperandrogénicos. J Clin Endocrinol Metab. (2008) 93:527–33. doi: 10.1210 / jc.2007-2053
PubMed Abstract | CrossRef Texto Completo | Google Scholar
17. Dewailly D, Vantyghem-Haudiquet MC, Sainsard C, Buvat J, Cappoen JP, Ardaens K, et al. Fenotipos clínicos y biológicos en deficiencia de 21-hidroxilasa de inicio tardío. J Clin Endocrinol Metab. (1986) 63:418–23. doi: 10.1210/jcem-63-2-418
PubMed Abstract | CrossRef Texto Completo | Google Scholar
18. Gönç EN, OzönA, Alikaşifoglu A, Engiz O, Bulum B, Kandemir N. (2011). Is basal serum 17-OH progesterone a reliable parameter to predict nonclassical congenital adrenal hyperplasia in premature adrenarche? Turk J Pediatr. 53:274–80.
PubMed Abstract | Google Scholar
19. Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2010) 95:4133–60. doi: 10.1210/jc.2009-2631
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Dacou-Voutetakis C, Dracopoulou M. High incidence of molecular defects of the CYP21 gene in patients with premature adrenarche. J Clin Endocrinol Metab. (1999) 84:1570–4. doi: 10.1210/jc.84.5.1570
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Ambroziak U, kepczynska-Eventos I, Kurylović, małunowicz EM, Wójcicka, miskević P, et al. The diagnosis of nonclassic congénita hiperplasia de la corteza suprarrenal due to 21-hydroxylase deficiency, based on sérico basal or post-ACTH stimulation 17-hydroxyprogesterone, can lead to false-positive diagnosis. Clin Endocrinol (Oxf). (2016) 84:23–9. doi: 10.1111 / precios.12935
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Ambroziak U, Kepczynska-I, Kurylovic, Wysłouch-Cieszyńska A, Małunowicz EM, Bartoszewicz, et al. LC-MS/MS improves screening towards 21-hydroxylase deficiency. Endocrinol Gynecol. (2015) 31:296–300. doi: 10.3109/09513590.2014.994599
PubMed Abstract | CrossRef Full Text | Google Scholar
23. BBC, CC, Goldman MM, Zhang K, Clark-N, Reitz re Welt CK. Medición simultánea de trece hormonas esteroideas en mujeres con síndrome de ovario poliquístico y mujeres de control mediante cromatografía líquida-espectrometría de masas en tándem. PLoS Uno. (2014) 9:e93805. doi: 10.1371 / journal.ponga.0093805
PubMed Abstract | CrossRef Texto Completo | Google Scholar
24. Ray JA, Kushnir MM, Yost RA, Rockwood AL, Wayne Meikle A. Mejora del rendimiento en la medición de 5 esteroides endógenos mediante LC-MS / MS combinada con espectrometría de movilidad iónica diferencial. Clin Chim Acta. (2015) 438:330–6. doi: 10.1016/j.cca.2014.07.036
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Stoupa A, González-Briceño L, Pinto G, Samara-Boustani D, Thalassinos C, Flechtner I, et al. Inadequate cortisol response to the tetracosactide (Synacthen®) test in non-classic congenital adrenal hyperplasia: an exception to the rule? Horm Res Paediatr. (2015) 83:262–7. doi: 10.1159/000369901
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. (1992) 90:584–95. doi: 10.1172/JCI115897
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Temeck JW, Pang SY, Nelson C, New MI. Defectos genéticos de esteroidogénesis en pubarquia prematura. J Clin Endocrinol Metab. (1987) 64:609–17. doi: 10.1210/jcem-64-3-609
PubMed Abstract | CrossRef Texto Completo | Google Scholar
28. Voutilainen R, Jääskeläinen J. Adrenarquia prematura: etiología, hallazgos clínicos y consecuencias. J Esteroide Bioquímico Mol Biol. (2015) 145:226–36. doi: 10.1016 / j. jsbmb.2014.06.004
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Oberfield SE, Sopher AB, Gerken AT. Approach to the girl with early onset of pubic hair. J Clin Endocrinol Metab. (2011) 96:1610–22. doi: 10.1210/jc.2011-0225
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Janus D, Wojcik M, Malunowicz E, Starzyk JB. Un caso de adherencias labiales recurrentes en un niño de 15 meses con hiperplasia suprarrenal congénita no clásica asintomática debido a deficiencia de 21-hidroxilasa. J Pediatr Endocrinol Metab. (2012) 25:1017–21. doi: 10.1515/jpem-2012-0190
PubMed Abstract | CrossRef Texto Completo | Google Scholar
31. Gopal-Kothandapani JS, Petkar A, O’Shea E, Banerjee I. Cabello perianal como presentación inusual de hiperplasia suprarrenal congénita no clásica. BMJ Case Rep. (2013) 2013:bcr2013010123. doi: 10.1136/bcr-2013-010123
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Falhammar H, Nordenström A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine. (2015) 50:32–50. doi: 10.1007/s12020-015-0656-0
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Chrousos GP, Loriaux DL, Mann DL, Cutler GB. Late-onset 21-hydroxylase deficiency mimicking idiopathic hirsutism or polycystic ovarian disease. Ann Intern Med. (1982) 96:143–8. doi: 10.7326/0003-4819-96-2-143
CrossRef Full Text | Google Scholar
34. Witchel SF. Non-classic congenital adrenal hyperplasia. Steroids. (2013) 78:747–50. doi: 10.1016/j.steroids.2013.04.010
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Carmina E, Rosato F, Jannì A, Rizzo M, Longo RA. Extensive clinical experience: relative prevalence of different androgen excess disorders in 950 women referred because of clinical hyperandrogenism. J Clin Endocrinol Metab. (2006) 91:2–6. doi: 10.1210/jc.2005-1457
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Moran C, Azziz R, Weintrob N, Witchel SF, Rohmer V, Dewailly D, et al. Resultado reproductivo de mujeres con hiperplasia suprarrenal no clásica con deficiencia de 21 hidroxilasa. J Clin Endocrinol Metab. (2006) 91:3451–6. doi: 10.1210 / jc.2006-0062
PubMed Abstract | CrossRef Texto Completo | Google Scholar
37. Levin JH, Carmina E, Lobo RA. ¿La secreción inadecuada de gonadotropinas de los pacientes con síndrome de ovario poliquístico es similar a la de los pacientes con hiperplasia suprarrenal congénita de inicio en adultos? Fertil Steril. (1991) 56:635–40. doi: 10.1016/S0015-0282(16)54592-0
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Carmina E, Lobo RA. Ovarian suppression reduces clinical and endocrine expression of late-onset congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Fertil Steril. (1994) 62:738–43. doi: 10.1016/S0015-0282(16)56998-2
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Ludwig E. Clasificación de los tipos de alopecia androgenética (calvicie común) que ocurren en el sexo femenino. Br J Dermatol. (1977) 97:247–54. doi: 10.1111 / j. 1365-2133. 1977.tb15179.x
PubMed Abstract | CrossRef Texto Completo | Google Scholar
40. Trakakis E, Papadavid E, Dalamaga M, Koumaki D, Stavrianeas N, Rigopoulos D, et al. Prevalencia de hiperplasia suprarrenal congénita no clásica debida a deficiencia de 21 hidroxilasa en mujeres griegas con acné: un estudio transversal basado en el hospital. J Eur Acad Dermatol Venereol. (2013) 27:1448–51. doi: 10.1111 / j. 1468-3083. 2012. 04613.x
PubMed Abstract | CrossRef Texto Completo | Google Scholar
41. Arlt W, Willis DS, Wild SH, Krone N, Doherty EJ, Hahner S, et al. Estado de salud de adultos con hiperplasia suprarrenal congénita: un estudio de cohorte de 203 pacientes. J Clin Endocrinol Metab. (2010) 95:5110–21. doi: 10.1210 / jc.2010-0917
PubMed Abstract | CrossRef Texto Completo | Google Scholar
42. Saygili F, Oge A, Yilmaz C. Hiperinsulinemia e insensibilidad a la insulina en mujeres con hiperplasia suprarrenal congénita no clásica debida a deficiencia de 21 hidroxilasa: la relación entre los niveles séricos de leptina y la hiperinsulinemia crónica. Horm Res. (2005) 63:270-4. doi: 10.1159/000086363
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Speiser PW, Serrat J, New MI, Gertner JM. Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1992) 75:1421–4. doi: 10.1210/jc.75.6.1421
PubMed Abstract | CrossRef Full Text | Google Scholar
44. Han TS, Stimson RH, Rees DA, Krone N, Willis DS, Conway GS, et al. Régimen de tratamiento con glucocorticoides y resultados de salud en adultos con hiperplasia suprarrenal congénita. Clin Endocrinol. (2013) 78:197–203. doi: 10.1111 / cen.Con sede en 12045
PubMed Abstract | CrossRef Texto Completo | Google Scholar
45. Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Uso de corticosteroides orales y riesgo de fracturas. J Bone Miner Res. (2000) 15:993-1000. doi: 10.1359 / jbmr.2000.15.6.993
PubMed Abstract | CrossRef Full Text | Google Scholar
46. Nebesio TD, Renbarger JL, Nabhan ZM, Ross SE, Slaven JE, Li L, et al. Differential effects of hydrocortisone, prednisone, and dexamethasone on hormonal and pharmacokinetic profiles: a pilot study in children with congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2016) 2016:17. doi: 10.1186/s13633-016-0035-5
PubMed Abstract | CrossRef Texto Completo | Google Scholar
47. Weintrob N, Dickerman Z, Sprecher E, Galatzer A, Pertzelan A. Deficiencia de 21 hidroxilasa no clásica en la infancia y la infancia: el efecto del tiempo de inicio de la terapia en la pubertad y la estatura final. Eur J Endocrinol. (1997) 136:188–95. doi: 10.1530 / eje.0.1360188
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Bonfig W, Bechtold S, Schmidt H, Knorr D, Schwarz HP. Reduced final height outcome in congenital adrenal hyperplasia under prednisone treatment: deceleration of growth velocity during puberty. J Clin Endocrinol Metab. (2007) 92:1635–9. doi: 10.1210/jc.2006-2109
PubMed Abstract | CrossRef Full Text | Google Scholar
49. Dörr HG. Crecimiento en pacientes con hiperplasia suprarrenal congénita clásica debido a deficiencia de 21-hidroxilasa. Horm Res. (2007) 68 (Suppl 5): 93-9. doi: 10.1159/000110587
PubMed Abstract | CrossRef Texto Completo | Google Scholar
50. Hoepffner W, Kaufhold A, Willgerodt H, Keller E. Los pacientes con hiperplasia suprarrenal congénita clásica debido a una deficiencia de 21-hidroxilasa pueden alcanzar su altura objetivo: la experiencia de Leipzig. Horm Res. (2008) 70:42-50. doi: 10.1159/000129677
PubMed Abstract | CrossRef Full Text | Google Scholar
51. van der Kaay D, van den Akker E. Ultralow-dose dexamethasone to preserve endogenous cortisol stress response in nonclassical congenital adrenal hyperplasia: a new promising treatment. Int J Endocrinol Metab. (2014) 12:e14657. doi: 10.5812/ijem.14657
CrossRef Full Text | Google Scholar
52. Lin-Su K, Vogiatzi MG, Marshall I, Harbison MD, Macapagal MC, Betensky B, et al. Treatment with growth hormone and luteinizing hormone releasing hormone analog improves final adult height in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. (2005) 90:3318–25. doi: 10.1210/jc.2004-2128
PubMed Abstract | CrossRef Full Text | Google Scholar
53. McCann-Crosby B, Chen M-J, Lyons SK, Lin Y, Axelrad M, Dietrich JE, et al. Nonclassical congenital adrenal hyperplasia: objetivos de tratamiento y transición. Pediatr Endocrinol Rev. (2014) 12:224-38.
PubMed Abstract | Google Scholar
54. Ogilvie CM, Crouch NS, Rumsby G, Creighton SM, Liao L-M, Conway GS. Hiperplasia suprarrenal congénita en adultos: una revisión de problemas médicos, quirúrgicos y psicológicos. Clin Endocrinol. (2006) 64:2–11. doi: 10.1111 / j. 1365-2265. 2005. 02410.x
PubMed Abstract | CrossRef Full Text | Google Scholar
55. Mah PM, Jenkins RC, Rostami-Hodjegan A, Newell-Price J, Doane A, Ibbotson V, et al. Weight-related dosing, timing and monitoring hydrocortisone replacement therapy in patients with adrenal insufficiency. Clin Endocrinol. (2004) 61:367–75. doi: 10.1111/j.1365-2265.2004.02106.x
PubMed Abstract | CrossRef Full Text | Google Scholar
56. Díaz A, Laufer Sr. Breech LLAA de PC en A, Care AC de O y GC en AH. Menstruación en niñas y adolescentes: uso del ciclo menstrual como signo vital. Pediatría. (2006) 118:2245–50. doi: 10.1542 / pediatría.2006-2481
CrossRef Texto Completo | Google Scholar
57. DP de Merke, DP de Poppas. Tratamiento de adolescentes con hiperplasia suprarrenal congénita. diabetes Endocrinol lancet. (2013) 1:341–52. doi: 10.1016/S2213-8587(13)70138-4
PubMed Abstract | CrossRef Full Text | Google Scholar
58. Verma S, Green-Golan L, VanRyzin C, Drinkard B, Mehta SP, Weise M, et al. Adrenomedullary function in patients with nonclassic congenital adrenal hyperplasia. Horm Metab Res. (2010) 42:607–12. doi: 10.1055/s-0030-1253385
PubMed Abstract | CrossRef Full Text | Google Scholar
59. New MI. Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2006) 91:4205–14. doi: 10.1210/jc.2006-1645
PubMed Abstract | CrossRef Full Text | Google Scholar
60. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2018) 103:4043–88. doi: 10.1210/jc.2018-01865
PubMed Abstract | CrossRef Full Text | Google Scholar
61. Kruse B, Riepe FG, Krone N, Bosinski HAG, Kloehn S, Partsch CJ, et al. Congenital adrenal hyperplasia – how to improve the transition from adolescence to adult life. Exp Clin Endocrinol Diabetes. (2004) 112:343–55. doi: 10.1055/s-2004-821013
PubMed Abstract | CrossRef Full Text | Google Scholar
62. Gastaud F, Bouvattier C, Duranteau L, Brauner R, Thibaud E, Kutten F, et al. Deterioro de los resultados sexuales y reproductivos en mujeres con formas clásicas de hiperplasia suprarrenal congénita. J Clin Endocrinol Metab. (2007) 92:1391–6. doi: 10.1210 / jc.2006-1757
PubMed Abstract | CrossRef Texto Completo | Google Scholar
63. Mercè Fernández-Balsells M, Muthusamy K, Smushkin G, Lampropulos JF, Elamin MB, Abu Elnour NO, et al. Uso prenatal de dexametasona para la prevención de la virilización en embarazos con riesgo de hiperplasia suprarrenal congénita clásica debido a la deficiencia de 21-hidroxilasa (CYP21A2): revisión sistemática y metanálisis. Clin Endocrinol. (2010) 73:436–44. doi: 10.1111 / j. 1365-2265. 2010. 03826.x
CrossRef Texto Completo | Google Scholar
64. Bidet M, Bellanné-Chantelot C, Galand – Portier MB, Golmard JL, Tardy V, Morel Y, et al. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2010) 95:1182–90. doi: 10.1210/jc.2009-1383
PubMed Abstract | CrossRef Full Text | Google Scholar
65. Birnbaum MD, Rose LI. Late onset adrenocortical hydroxylase deficiencies associated with menstrual dysfunction. Obstet Gynecol. (1984) 63:445–51.
PubMed Abstract | Google Scholar
66. Cuhaci N, Aydin C, Yesilyurt A, Pinarli FA, Ersoy R, Cakir B. Nonclassical congenital adrenal hyperplasia and pregnancy. Case Rep Endocrinol. (2015) 2015:296924. doi: 10.1155/2015/296924
PubMed Abstract | CrossRef Full Text | Google Scholar
67. Deneux C, Tardy V, Anne D, Mornet E, Billaud L, Charron D, et al. Correlación fenotipo-genotipo en 56 mujeres con hiperplasia suprarrenal congénita no clásica debida a deficiencia de 21-hidroxilasa. J Clin Endocrinol Metab. (2001) 86:207–13. doi: 10.1210 / jcem.86.1.7131
PubMed Abstract | CrossRef Texto Completo | Google Scholar
68. Simonetti L, Bruque CD, Fernández CS, Benavides-Mori B, Delea M, Kolomenski JE, et al. Actualización de mutaciones en CYP21A2: Análisis exhaustivo de bases de datos y variantes genéticas publicadas. Hum Mutat. (2018) 39:5–22. doi: 10.1002 / humu.23351
PubMed Abstract | CrossRef Full Text | Google Scholar
69. OMIM Entry – * 613815 – CYTOCHROME P450 FAMILY 21 SUBFAMILY A POLYPEPTIDE 2; CYP21A2.
Google Scholar
70. Miller WL, Witchel SF. Prenatal treatment of congenital adrenal hyperplasia: risks outweigh benefits. Am J Obstet Gynecol. (2013) 208:354–9. doi: 10.1016/j.ajog.2012.10.885
PubMed Abstract | CrossRef Full Text | Google Scholar
71. Lekarev O, New MI. Adrenal disease in pregnancy. Best Pract Res Clin Endocrinol Metab. (2011) 25:959–73. doi: 10.1016/j.beem.2011.08.004
PubMed Abstract | CrossRef Full Text | Google Scholar
72. New MI, Abraham M, Yuen T, Lekarev O. An update on prenatal diagnosis and treatment of congenital adrenal hyperplasia. Semin Reprod Med. (2012) 30:396–9. doi: 10.1055/s-0032-1324723
PubMed Abstract | CrossRef Full Text | Google Scholar
73. Kazmi D, Bailey J, Yau M, Abu-Amer W, Kumar A, Low M, et al. New developments in prenatal diagnosis of congenital adrenal hyperplasia. J Steroid Biochem Mol Biol. (2017) 165:121–3. doi: 10.1016/j.jsbmb.2016.06.016
PubMed Abstract | CrossRef Full Text | Google Scholar
74. Clayton PE, Miller WL, Oberfield SE, Ritzén EM, Sippell WG, Speiser PW, et al. Declaración de consenso sobre la deficiencia de 21-hidroxilasa de la Sociedad Europea de Endocrinología Pediátrica y la sociedad endocrina pediátrica lawson wilkins. Horm Res. (2002) 58: 188-95. doi: 10.1159/000065490
PubMed Abstract | CrossRef Texto Completo | Google Scholar
75. Nandagopal R, Sinaii N, Avila NA, Van Ryzin C, Chen W, Finkielstain GP, et al. Perfil fenotípico de padres con hiperplasia suprarrenal congénita críptica no clásica: hallazgos en 145 familias no emparentadas. Eur J Endocrinol. (2011) 164:977–84. doi: 10.1530/EJE-11-0019
PubMed Abstract | CrossRef Texto Completo | Google Scholar
76. Takasu N, Nakachi K, Higa H. El desarrollo de hipertiroidismo de Graves causó una crisis suprarrenal en un paciente con deficiencia de 21 hidroxilasa no clásica no reconocida previamente. Médico en prácticas. (2010) 49:1395–400. doi: 10.2169 / medicina interna.49.3573
PubMed Abstract | CrossRef Full Text | Google Scholar
77. Meyer-Bahlburg HFL, Dolezal C, Baker SW, Carlson AD, Obeid JS, New MI. Prenatal androgenization affects gender-related behavior but not gender identity in 5-12-year-old girls with congenital adrenal hyperplasia. Arch Sex Behav. (2004) 33:97–104. doi: 10.1023/B:ASEB.0000014324.25718.51
CrossRef Full Text | Google Scholar
78. Meyer-Bahlburg HFL, Dolezal C, Baker SW, New MI. Orientación sexual en mujeres con hiperplasia suprarrenal congénita clásica o no clásica en función del grado de exceso prenatal de andrógenos. Arch Sex Behav. (2008) 37:85–99. doi: 10.1007 / s10508-007-9265-1
Resumen de PubMed / Texto completo cruzado / Google Scholar