- Definitionen und Prävalenz
- Diagnose
- NCCAH-Manifestationen bei Frauen
- Manifestationen in der Kindheit
- Manifestationen im Jugend- und Erwachsenenalter
- NCCAH-Management von der ersten Manifestation bis zum Erwachsenenalter
- Behandlung im Kindesalter
- Management während der Adoleszenz
- Patienten, die seit der Kindheit behandelt wurden
- Frauen mit erster Manifestation während der Adoleszenz oder hyperandrogenen Symptomen nach Absetzen der Behandlung
- NCCAH und Reproduktion
- Subfertilität
- Fehlgeburten
- Fruchtbarkeitsplanung
- Pränatale Behandlung von CAH
- Behandlung während der Schwangerschaft
- Weitere Sonderthemen
- Stressmanagement bei NCCAH
- Psychobiologische Aspekte im NCCAH
- Schlussfolgerungen
- Autorenbeiträge
- Erklärung zum Interessenkonflikt
Definitionen und Prävalenz
Die kongenitale Nebennierenhyperplasie (CAH) umfasst eine Familie von autosomal-rezessiven Erkrankungen, die durch eine leichte bis akut beeinträchtigte Cortisolsynthese aufgrund eines Mangels an einem der fünf für die Cortisolproduktion erforderlichen steroidogenen Nebennierenenzyme gekennzeichnet sind (1, 2). Herkömmlicherweise wird CAH in (a) klassische (CCAH) unterteilt, die sich mit Salzverschwendung oder der einfachen virilisierenden Form präsentiert, die sich bei der Geburt und / oder in der Neugeborenenperiode manifestiert, und (b) nicht-klassische (NCCAH), die eine weniger schwere Form der Störung darstellt, die keine genitale Ambiguität aufweist, nicht sofort lebensbedrohlich ist und später im Leben auftritt oder asymptomatisch bleibt oder als andere Krankheit falsch diagnostiziert wird (3). Die Grenzen zwischen den verschiedenen Formen von CAH sind jedoch nicht genau definiert, was die mit dieser Störung verbundenen Herausforderungen tendenziell erhöht. Es ist daher ratsam, CAH als ein Kontinuum von Phänotypen zu betrachten, von schwer bis leicht oder auch asymptomatisch (3).Die häufigste Form von CAH wird durch einen 21-Hydroxylase-Mangel (21OHD) verursacht, der zu einer beeinträchtigten oder keiner Umwandlung von 17 Hydroxyprogesteron (17 OHP) in 11 Desoxycortisol und von Progesteron in Desoxycorticosteron führt. Die Blockade der Steroidumwandlung führt zu einer erhöhten Produktion von Androgenvorläufern unter CRH-ACTH-Stimulation, was zu biochemischem Hyperandrogenismus führt, der durch erhöhte 17-OHP-Spiegel gekennzeichnet ist. Die klassische Form der Krankheit tritt bei 1 von 16.000 Lebendgeburten weltweit auf. Es sollte jedoch erwähnt werden, dass die wahrgenommene Inzidenz der Krankheit mit der verwendeten Screening-Methode zusammenhängt. Beispielsweise wurde berichtet, dass die Inzidenz der klassischen Form von CAH in Schweden bei Verwendung eines Fallumfrageansatzes 1: 11.500 beträgt, diese Zahl sinkt jedoch auf 1: 9.800, wenn ein hormonelles Screening angewendet wird. Ebenso liegt die Inzidenz in Frankreich und der Schweiz zwischen 1:15.472 und 1:23.000 und 1:10.749 und 1:11.661, wenn die verschiedenen Screening-Methoden verwendet werden (4).NCCAH ist viel häufiger und tritt bei etwa 1 von 1.000 Kaukasiern und häufiger bei bestimmten ethnischen Gruppen wie aschkenasischen Juden (1:27), Hispanics (1:53), Jugoslawen (1:62) und Italienern auf (1:300) (5, 6). Nichtsdestotrotz fehlen Daten zur Prävalenz von NCCAH basierend auf verschiedenen Schätzmethoden (Fallstudie, hormonelles Screening und molekulare Tests). NCCAH gilt als die häufigste autosomal-rezessive endokrine Störung mit einer Trägerfrequenz von 1:25 bis 1:10. In NCCAH aufgrund von 21-OHD wird die verbleibende enzymatische Aktivität auf etwa 10-70% geschätzt (7-9).
Diagnose
Im Vergleich zur Diagnose der klassischen Form der Krankheit, die bei der Geburt oder während der Neugeborenenperiode aufgrund von Genital-Ambiguität und / oder salzverschwendenden Symptomen oder durch in einigen Ländern eingesetzte Screening-Programme gestellt wird, sind die meisten Fälle von NCCAH nicht leicht nachweisbar (4, 10). Darüber hinaus bleiben viele Personen im Kindes- und Jugendalter asymptomatisch, haben eine normale Fortpflanzungsfunktion und werden erst durch die Diagnose eines anderen Familienmitglieds und anschließende Tests auf NCCAH aufmerksam (11). Die meisten Frauen mit NCCAH suchen jedoch medizinische Hilfe auf, wenn Symptome eines Androgenüberschusses auftreten, und wenn der klinische Verdacht zum Testen auffordert, weisen erhöhte basale 17-OHP-Spiegel höchstwahrscheinlich auf eine Diagnose von NCCAH hin.Tatsächlich empfehlen die von der Endocrine Society vorgeschlagenen klinischen Richtlinien einen nicht stimulierten Ausgangswert von 17 OHP als Screening-Tool für NCCAH. Morgen 17 OHP-Spiegel >6 nmol / L in der Follikelphase bei menstruierenden Frauen sollten eine weitere Bewertung veranlassen, da gezeigt wurde, dass Spiegel über 6 nmol / L 90% der NCCAH-Individuen erfassen (12, 13). Zufällige Messungen von 17 OHP haben sich nicht als hilfreich erwiesen, da diese bei Patienten mit NCCAH häufig normale Werte ergeben und darüber hinaus in der Lutealphase bei der Hälfte der gesunden Frauen extrem hoch sind (13). In unseren Daten von 280 Patienten mit der Krankheit wurden jedoch sechs Patienten (2.1%) hatte einen Ausgangswert von 17 OHP < 6 nmol/L (14). Darüber hinaus Bidet et al., in einer großen Kohorte von Frauen mit NCCAH, die durch molekulare Techniken verifiziert wurden, fanden auch basale 17 OHP-Werte niedriger als 6 nmol / L in 8% der untersuchten Probanden (15). Schließlich, basierend auf Daten von Speiser et al., 9% von Personen mit NCCAH zeigten 17 OHP-Werte niedriger als 2 ng / ml (das entspricht 6nmol / l). Nach anderen Studien ist ein Ausgangswert von 17 OHP zwischen 5,1 und 9 nmol / L ausreichend für die Diagnose von NCCAH (13, 16, 17). Vor kurzem ein Niveau von basalen 17 OHP von 4.6 nmol / l wurde als Schwellenwert für ACTH-Tests vorgeschlagen, um NCCAH bei Probanden mit vorzeitiger Adrenarche im Kindesalter vorherzusagen (18).
Die Summe dieser Befunde und Vorschläge zeigt, dass die Auswahl der Patienten, die sich einem Synakthen-Stimulationstest unterziehen, von Fall zu Fall bewertet werden sollte. Für die Diagnose ist ein 17-OHP-Poststimulationspegel über 3 nmol / l erforderlich (19). Heterozygoten, die eine CYP21A2-Mutation tragen, weisen nach ACTH-Stimulation leicht erhöhte 17-OHP-Spiegel auf, obwohl es bei nicht betroffenen Probanden zu Überschneidungen kommt (9). Wie Dacou-Voutetakis et al. haben darauf hingewiesen, wenn die Summe der basalen und Post-Stimulation 17 OHP-Werte 1,5 nmol / L überschreitet, dann ist die Möglichkeit der Heterozygotie außergewöhnlich hoch (20).
Eine weitere wichtige Überlegung betrifft die Techniken, die für die 17-OHP-Bewertung verwendet werden. 17 OHP-Spiegel werden durch eine Vielzahl von Immunoassay-Methoden gemessen, aber wie kürzlich gezeigt wurde, wurden die genauesten und zuverlässigsten Ergebnisse durch die Implementierung der Kombination von Flüssigkeitschromatographie mit Massenspektrometrie (LC-MS / MS) erreicht. In der Tat wurden viele falsch positive 17-OHP-Messungen gefunden, als LC-MS / Ms-Messungen mit Standardmethoden verglichen wurden (21) Letztere Verfahren werden jedoch nicht universell verwendet, im Falle einer unsicheren Diagnose liefern sie genauere Ergebnisse. Bemerkenswert ist, dass die Screening- und Diagnoseschwellen für LC-MS / MS quantifiziert 17 OHP müssen noch definiert werden. Diese Schwellenwerte sind wahrscheinlich niedriger als bei Immunoassays aufgrund der erhöhten Spezifität von LC-MS / MS, einer Methode, die weniger anfällig für Kreuzreaktivität und Interferenzen ist (22, 23). Ob für die endgültige Diagnose ein Steroidprofil im Urin erforderlich ist, muss noch geklärt werden (22). In Grenzfällen ist es ratsam, nach dem ACTH-Stimulationstest ein vollständiges adrenokortikales Profil zu erhalten, um einen 21-Hydroxylase-Mangel von anderen Enzymdefekten zu unterscheiden und eine feste Diagnose zu stellen.Insbesondere sollte in zweideutigen Fällen ein vollständiges Steroidprofil durchgeführt werden, um einen 21-Hydroxylase-Mangel zu bestätigen und andere enzymatische Veränderungen auszuschließen. Die Aufnahme von 17 OHP, Cortisol, 11-Desoxycorticosteron, 11-Desoxycortisol, 17-OH-Pregnenolon, Dehydroepiandrosteron und Androstendion in ein Serumsteroidprofil wäre nützlich, um andere Ursachen für CAH auszuschließen, wie z. B. 11β-Hydroxylase-Mangel und seltener 3β-Hydroxysteroid-Dehydrogenase-Mangel oder P450-Oxidoreduktase-Mangel (24). Während Gentests derzeit nicht als primäres diagnostisches Instrument für NCCAH angesehen werden, sind sie für die Diagnosebestätigung und für die genetische Beratung obligatorisch (3).
Was die Cortisolspiegel betrifft, wird im Allgemeinen ein Poststimulationswert von mindestens 496 nmol/L erwartet. Bemerkenswert ist, dass Stoupa et al., 60% von 47 Kindern mit NCCAH infolge von 21 OHD hatten niedrige Cortisolwerte nach der Stimulation, ein Befund, der auf die Notwendigkeit einer verstärkten Überwachung der Entwicklung einer Nebenniereninsuffizienz während schwerer Stressor-Ereignisse hinweist (25).
NCCAH-Manifestationen bei Frauen
Die klinische Expression von NCCAH ist durch einen hohen Polymorphismus gekennzeichnet, der nicht nur das Erkrankungsalter, sondern auch die verschiedenen Anzeichen und Symptome betrifft. Es wird berichtet, dass das erste klinische Auftreten von NCCAH in 11% der Fälle vor dem Alter von 10 Jahren und in 80% zwischen dem Alter von 10 und 40 Jahren auftritt (12). Die Genotyp-Phänotyp-Korrelation in CAH und NCCAH ist noch nicht aufgeklärt. Speiser et al. deuten darauf hin, dass die meisten, aber nicht alle der phänotypischen Variabilität in 21-Hydroxylase-Mangel resultiert aus allelischen Variation in CYP21A2 (26).
Manifestationen in der Kindheit
In der Neugeborenenperiode bleiben typischerweise NCCAH-Frauen asymptomatisch und haben normale äußere Genitalien. Der früheste gemeldete Fall von NCCAH ist ein 6 Monate altes Mädchen, das Schamhaare entwickelte (11). In der Regel beginnen klinische Befunde und Symptome in NCCAH-Fällen ab dem Alter von 5 Jahren oder sogar später und hängen mit erhöhten Androgenspiegeln zusammen, obwohl in einigen Fällen auch ein leichter Cortisolmangel auftreten kann (8). In 92% der NCCAH-Fälle tritt als erstes Symptom eine vorzeitige Pubarche (PP) auf, die bei Mädchen unter 8 Jahren und bei Jungen unter 9 Jahren auftritt (12) mit einer geschätzten Prävalenz zwischen 10 und 11, 3% (15). Bemerkenswert ist, dass bei Kindern mit PP nach verschiedenen Studien die Inzidenz von NCCAH zwischen 5 und 30% liegt (20, 27). Basierend auf unserer Analyse von 280 Patienten mit molekularer Bestätigung von NCCAH stellten wir jedoch fest, dass die Inzidenz von PP bei 94 Frauen unter 8 Jahren bis zu 88% betrug. Andererseits identifizierte eine Analyse von 45 Männern mit NCCAH PP nur bei 29% der Probanden (14). Darüber hinaus sollten wir bedenken, dass etwa die Hälfte der Probanden mit PP heterozygote Träger einer CYP21A2-Mutation sein kann.
Ein weiterer Aspekt, der geklärt werden muss, ist die Beziehung zwischen PP und vorzeitiger Pubertät. Es wird vermutet, dass in einigen Fällen die periphere Aromatisierung von Nebennierenandrogenen zu Östrogenen die Hypothalamus-Hypophysen-Gonaden-Achse aktivieren kann, was zu einer sekundären vorzeitigen Pubertät führt (28). In einem solchen Fall ist eine regelmäßige Nachsorge erforderlich. Einige Kinder entwickeln Hirsutismus, Akne und / oder schnelles Wachstum und manifestieren hohe Struktur und Knochenalter Fortschritt (29). Letzteres kann zu einer verkürzten Endhöhe als Folge einer schnellen Epiphysenfusion führen. Andere seltene klinische Befunde im Kindesalter sind labiale Adhäsion, perianales Haar, Klitoromegalie und Heiserkeit der Stimme (30-32).
Manifestationen im Jugend- und Erwachsenenalter
Während der Adoleszenz und des Erwachsenenalters weist NCCAH bei Frauen hyperandrogene Symptome auf, die dem polyzystischen Ovarialsyndrom (PCOS) ähneln (33). Zu den Symptomen gehören Hirsutismus, Akne, androgene Alopezie, Anovulation, Menstruationsstörungen und Unfruchtbarkeit. In einer multizentrischen Studie waren die häufigsten Symptome bei jugendlichen und erwachsenen Frauen Hirsutismus (59%), Oligomenorrhoe (54%) und Akne (33%). Bei 161 Frauen mit NCCAH zeigten sich Hirsutismus (78%), Menstruationsstörungen (54,7%) und verminderte Fruchtbarkeit (12%) (34). In unserer Gruppe von 161 Frauen mit der Krankheit zeigten 63% der Patientin einen polyzystischen ovarialähnlichen Phänotyp (14). Dieser Befund hat eine umgekehrte Implikation, da die Inzidenz von NCCAH bei Frauen mit Hyperandrogenämie hoch ist. Sicherlich ist die Studie von Witcel et al. die Meldung einer NCCAH-Inzidenz von 1 bis 33% bei hyperandrogenen Frauen ist von großem Interesse. In einer anderen Studie mit 950 Frauen, die zur Beurteilung des Androgenüberschusses überwiesen wurden, wurde bei 4,2% von ihnen NCCAH festgestellt. Die NCCAH-Patienten waren jedoch jünger und behaarter als die anderen Gruppen hyperandrogener Patienten. Sie waren auch durch signifikant höhere Testosteronspiegel, freies Testosteron und 17 OHP gekennzeichnet als Patienten mit anderen hyperandrogenen Syndromen. Im Eierstock-Ultraschall zeigten 77% von ihnen polyzystische Eierstöcke und 41% erhöhte Eierstockgröße. Insgesamt erfüllten sie die PCOS-Kriterien nach NIH und Rotterdam mit einem Prozentsatz von 56 bzw. 72,8% (35).
Die fortschreitende Natur der Krankheit wird durch die Tatsache unterstrichen, dass die Prävalenz von Hirsutismus mit zunehmendem Alter zunimmt und vor der Pubertät als selten beobachtet wurde. Hirsutismus ist die häufigste klinische Manifestation bei Patienten mit NCCAH, die zwischen 71 und 96% liegt (28, 36). Pubertäre Mädchen mit NCCAH weisen typischerweise Hirsutismus auf (37, 38). Am anderen Ende des Spektrums ist das Problem der Alopezie. Männliche Glatzenbildung wird bei NCCAH-Patienten berichtet. Die Prävalenz von Alopezie schien ebenfalls mit zunehmendem Alter von 6% bei Patienten im zweiten Lebensjahrzehnt auf 19% im fünften Lebensjahrzehnt zuzunehmen, was wiederum auf die fortschreitende Natur der Krankheit hinweist (39). Akne wird bei fast 33% der NCCAH-Patienten berichtet (36). Bemerkenswerterweise wurden Mutationen des CYP21A2-Gens, die NCCAH verursachten, bei 4,9% von 123 erwachsenen Frauen mit schwerer Akne nachgewiesen (40).Menstruationsunregelmäßigkeiten, einschließlich Oligomenorrhoe oder sekundärer Amenorrhoe, können häufig das präsentierende Zeichen von NCCAH bei Personen nach der Menarche sein. Darüber hinaus leiden 8-9% der Fälle auch an primärer Amenorrhoe und suchen aus diesem Grund zum ersten Mal einen Arzt auf. Darüber hinaus ist Oligomenorrhoe bei NCCAH-Individuen häufiger als primäre Amenorrhoe. Zum Beispiel in einer Reihe von Moran et al. bei NCCAH-Frauen im Alter von 10 bis 19 Jahren war Oligomenorrhoe in 56% der Fälle im Vergleich zu nur 9% der Adoleszenz mit primärer Amenorrhoe vorhanden (12). Zusammenfassend lässt sich sagen, dass so viele NCCAH-Symptome denen von PCOS ähneln, dass es nicht verwunderlich ist, dass NCCAH als der große Nachahmer von PCOS bezeichnet wurde. In jedem Fall sollte eine Diagnose von NCCAH bei der Beurteilung einer jungen Frau in Betracht gezogen werden, die wegen hyperandrogener Symptome überwiesen wird.
In einer Studie von Arlt et al., Patienten mit NCCAH wurden durch bedeutsam höheren BMI und niedrigere Endhöhe im Vergleich zu übereinstimmenden Kontrollen charakterisiert (41). Diese Daten deuten in Verbindung mit einigen Studien, die auf eine Insulinunempfindlichkeit hinweisen, auf ein ungünstiges Stoffwechselprofil hin, das pathophysiologisch durch die Wirkung erhöhter Androgenkonzentrationen und / oder als Folge einer Glukokortikoidtherapie erklärt werden könnte (42, 43). Osteoporose kann auch bei diesen Probanden nachgewiesen werden, wahrscheinlich als Folge der Kortikosteroidtherapie (34).
NCCAH-Management von der ersten Manifestation bis zum Erwachsenenalter
Sobald die Diagnose von NCCAH gestellt wurde, sind mehrere Fragen im Zusammenhang mit nachfolgenden Behandlungsempfehlungen zu berücksichtigen. Die erste zu behandelnde Frage ist, ob eine Glucocorticoid (GC) -Therapie indiziert ist. Die allgemeine Überlegung hinter diesem Ansatz ist, dass durch die Bereitstellung ausreichender Cortisolkonzentrationen für die Patientin ihr täglicher Bedarf gedeckt wird und folglich die Stimulation der CRH-ACTH-Achse verjüngt wird, was zu einer verminderten Androgenproduktion der Nebennieren führt. Die allgemeine Regel für diesen Ansatz ist, dass GCs in Ersatz- und nicht in pharmakologischen Dosen verabreicht werden, während der Einfluss von Alter, Geschlecht, Labordaten, patientenspezifischen Empfehlungen und Behandlungszielen auf die Glukokortikoid-Ersatztherapie ernsthaft berücksichtigt wird. Wir sollten uns jedoch auch bewusst sein, dass eine verlängerte GC-Substitutionstherapie zu Hyperkortisolämie führen kann, mit den daraus resultierenden gut dokumentierten negativen Auswirkungen auf jeden Aspekt des Stoffwechsels, insbesondere Wachstum, Fettverteilung und Insulinresistenz sowie auf psychologisches Profil. Ein weiterer wesentlicher Nachteil dieses Ansatzes ist das Fehlen eines adäquaten klinischen Index oder biochemischen Markers für eine adäquate Ersatzdosis, wie sie in Bezug auf TSH-Werte bei Hypothyreose bestehen.Schließlich erschweren die Tatsache, dass es keinen Konsens darüber gibt, welche GC verwendet werden sollte, und das Fehlen von Langzeitdaten zu verschiedenen Modalitäten der GC-Verabreichung die Entscheidungen der behandelnden Ärzte und die Auswahl der optimalen Wahl für jeden Patienten. Hydrocortison wird typischerweise bei Kindern angewendet, da es dem natürlichen Hormon (Cortisol) am ähnlichsten ist, bei Jugendlichen und jungen Frauen jedoch aufgrund der Notwendigkeit einer mehrfachen täglichen Dosierung nicht als geeigneter Ansatz angesehen wird. Daher bevorzugen die meisten erwachsenen Endokrinologen entweder Dexamethason oder Prednisolon in geeigneten Dosen. Andererseits muss darauf hingewiesen werden, dass die Äquivalenz verschiedener GCs auf ihrer entzündungshemmenden Wirkung und nicht auf verschiedenen Aspekten des menschlichen Stoffwechsels beruht. Da Cortisol fast 20% des menschlichen Genoms betrifft, werden unterschiedliche Reaktionen verschiedener GCs in verschiedenen Geweben erwartet. Diese Wahrnehmung wird durch die Tatsache bestätigt, dass die Verabreichung von Dexamethason an Patienten mit CAH eine Verschlechterung der Insulinresistenzindizes im Vergleich zu anderen GCs gezeigt hat (44). Darüber hinaus übt die Verabreichung von 2,5–7,5 mg Prednisolon, eine als normal angesehene Dosis, einen langjährigen negativen Einfluss auf den Knochenstoffwechsel aus (45). Daher sollte Hydrocortison als die beste Behandlungsform bei GC-Supplementationstherapie angesehen werden. Das Aufkommen der neu synthetisierten Hydrocortisonformel mit einer Pille pro Tag und ihre ersten positiven Ergebnisse bei Patienten mit CAH sind vielversprechend für die Zukunft (46).
Behandlung im Kindesalter
NCCAH-Patienten, bei denen im Kindesalter Anzeichen von PP diagnostiziert werden, können mit Hydrocortison behandelt werden, um die Nebennierenhormone zu unterdrücken und ein schnelles Fortschreiten des Knochenalters zu verhindern, das die endgültige Körpergröße beeinflussen könnte. Bei den Patienten, bei denen die Hydrocortisonbehandlung 1 Jahr vor Beginn der Pubertät begonnen wurde und die ein Knochenalter unter 9 Jahren hatten, blieb die endgültige Größe innerhalb des genetischen Potenzials (47). Verfügbare Studien weisen darauf hin, dass sich die Körpergröße bei Erwachsenen der erwarteten Zielgröße bei Patienten näherte, die engmaschig überwacht wurden und die Medikationspläne strikt einhielten (48-50). Eine kürzlich durchgeführte kleine Studie mit fünf Fällen (Patienten im Alter von 6, 1 bis 9, 2 Jahren) zeigte, dass eine ultraniedrige Dosis Dexamethason eine vielversprechende Option zur Verringerung von endogenem Stress und seinen Auswirkungen ist. Ob dies ein realistischer Ansatz ist und wie hoch die verabreichte Dosis sein sollte, muss noch geklärt werden (51). Eine Studie von Nebesio et al. vergleich der hormonellen Wirkungen und Pharmakokinetik von Hydrocortison, Prednisolon und Dexamethason bei 9 präpubertären Patienten mit CCAH. Sie zeigten, dass Dexamethason wirksamer war als die anderen Formen, um signifikant niedrigere Nebennierenhormonspiegel zu erreichen, was darauf hindeutet, dass Dexamethason wirksamer für die Unterdrückung der Nebennierenandrogenproduktion ist (46). Natürlich sind weitere Studien erforderlich, um diesen Befund zu überprüfen oder abzulehnen. Darüber hinaus können in einigen Fällen erhöhte Androgenkonzentrationen zu einer sekundären Stimulation der GnRH-Achse führen, was zu einer vorzeitigen Pubertät führt. In einem solchen Fall kann ein Parallelkurs mit GnRH-Analog vorgeschrieben werden, wenn das Knochenalter signifikant höher als das chronologische Alter ist und / oder die projizierte Endhöhe in keinem Verhältnis zur Zielhöhe steht.
Eine weitere wichtige Überlegung betrifft Fälle von gleichzeitigem GH-Mangel für den Fall, dass die Behandlung mit GnRH-Analoga und GH-Verschreibung zur Erreichung der Zielgröße führte (52). Gemäß den aktuellen Richtlinien wird die oben beschriebene Therapie nur in den Fällen vorsichtig empfohlen, in denen die vorhergesagte Höhe SD -2,25 < die Zielhöhe oder sogar niedriger ist (53). Eine alternative Indikation für den Beginn einer Hydrocortisonbehandlung ist eine unzureichende Cortisolreaktion nach ACTH-Stimulation (36).
Die bevorzugte GC-Behandlung bei Kindern ist in der Regel Hydrocortison 10-15 mg/m2, aufgeteilt in drei Dosen. Häufig haben sich auch niedrigere Dosen ab 6 mg / m2 / Tag als wirksam erwiesen (34). Eine Überdosierung sollte vermieden werden, da dies zu schlechtem Wachstum und cushingoiden Merkmalen führen kann. Regelmäßiges Wachstumsmuster, ein mit dem chronologischen Alter kompatibles Knochenalter und das Fehlen einer zentralen Adipositas können ebenfalls als klinische Indizes für ein angemessenes Management dienen. In Bezug auf das biochemische / hormonelle Profil gelten Androstendion- und Testosteronspiegel im mittleren bis oberen Bereich für das Knochenalter als bessere Marker für eine adäquate GC-Ersatztherapie bei Kindern mit NCCAH. Unterdrückte Testosteronspiegel wurden jedoch bei 10% der NCCAH-Patienten gefunden, während weitere 28% der Patienten erhöhte Testosteronkonzentrationen aufwiesen, wobei dieses Phänomen möglicherweise auf die Hydrocortisonvariabilität zurückzuführen war (54, 55). DHEAS-Werte werden mit sehr kleinen Dosen verringert, während die Unterdrückung von 17 OHP- und Progesteronspiegeln sehr hohe GC-Dosen erfordert. Es ist zu beachten, dass 17 OHP–Werte für die Diagnose entscheidend sind, aber während der Nachsorge nicht hilfreich sind. Es ist zu beachten, dass die Blutentnahme zur hormonellen Beurteilung ohne Absetzen der Therapie durchgeführt werden muss. Aufgrund der Verwirrung der Krankheit und ihrer Vielschichtigkeit gibt es jedoch keine spezifischen Richtlinien für den Zeitpunkt von Regimeänderungen oder die Beendigung der Glukokortikoidtherapie bei Kindern.
Management während der Adoleszenz
Patienten, die seit der Kindheit behandelt wurden
Bis zur Etablierung des normalen Menstruationsmusters bei NCCAH-Mädchen wird die Fortsetzung der GCs, die im Kindesalter begann, dringend empfohlen (56). Jugendliche Patienten zeigen jedoch häufig keine ausreichende Compliance mit der chronischen Verabreichung von Arzneimitteln und lassen häufig Dosen aus. Zusätzlich während der Pubertät, fällt die Halbwertszeit von Hydrocortison durch 50% infolge der erhöhten Niveaus IGF-1, die Tätigkeit 11ßOHSD verringert, sowie wegen der erhöhten Kortisolabfertigung, die von der Verstärkung der glomerulären Filtrationsrate (57) sich ergibt. Bei der jungen Frau, 2 Jahre nach der Menarche und wenn normale Ovulationszyklen aufgezeichnet wurden, wird ein patientenzentrierter Ansatz für die möglicherweise auftretenden hyperandrogenen Symptome dringend empfohlen.
Bei schweren hyperandrogenen Befunden wie Hirsutismus, Akne und / oder Oligomenorrhoe wird eine Fortsetzung der Behandlung in Betracht gezogen. Es ist wichtig, sich an die zerbrechlichen Empfindlichkeiten eines jungen Jugendlichen zu erinnern, die durch die „abweisenden“ Anzeichen von Hyperandrogenismus ernsthaft geschädigt werden. Die Kombination von hyperandrogenen Anzeichen, Menstruationsstörungen und schlechter Lebensqualität ist gut dokumentiert und bei jüngeren Frauen besonders hoch. Ein weiterer zu berücksichtigender Punkt ist die Reaktion der Nebennierenreserve nach ACTH-Stimulation. Da das maximale Cortisol von pubertären und erwachsenen Frauen nach Stimulation unter 496 nmol / l liegt, sollte bei Stress eine Steroidbehandlung verabreicht werden (58). Wenn andererseits bei dem jungen Patienten keines der oben genannten Symptome auftritt, kann die GC-Behandlung abgebrochen werden.
Frauen mit erster Manifestation während der Adoleszenz oder hyperandrogenen Symptomen nach Absetzen der Behandlung
Bei Vorliegen hyperandrogener Symptome wird dringend ein patientenorientierter Ansatz empfohlen, der sich auf die Hauptbeschwerden des Patienten konzentriert. Dies ist weitaus besser als ein allgemeiner universeller Modus operandi, da bei letzterem Ansatz der junge Patient enttäuscht sein wird, was zum Abbruch der Behandlung und infolgedessen zu nachteiligen gesundheitlichen Folgen im Erwachsenenalter führt. In Fällen mit schwerem Hirsutismus ist ein Kurs von 6 bis 12 Monaten mit oralen Kontrazeptiva (OCPs) ein vernünftiges Verfahren angesichts der positiven Auswirkungen von OCPs auf die SHBG-Leberproduktion und die Abnahme der Androgenfreisetzung aus dem Eierstock. Eine klinische Verbesserung wird nach mindestens 2-3 Monaten OCPs-Initiierung erwartet, während die gleichzeitige Anwendung eines Gestagens mit minimalen androgenen Eigenschaften dringend empfohlen wird. Wenn OCPs als First-Line-Ansatz versagen, können Antiandrogene (Spironolacton, Flutamid, Bicalutamid, Cyproteronacetat und Finasterid) zur Behandlung hinzugefügt werden. Nach unserer Erfahrung hat die Verabreichung von Bicalutamid bei schwerer Akne eine signifikante Verbesserung erzielt, bei schwerem Hirsutismus wurden jedoch keine ähnlichen Ergebnisse erzielt. Kosmetische Ansätze wie Laseranwendung und Enthaarungsmittel können auch für Frauen vorgeschlagen werden, die über übermäßiges oder unerwünschtes Haarwachstum oder Kopfhaarverlust (androgene Alopezie) klagen (32, 34, 57).
Bei Patienten mit unzureichender Cortisolsekretion nach Stimulation oder wenn OCPs und Antiandrogene nicht vertragen werden können, wird ein Kurs mit GCs dringend empfohlen (57). Daten von New et al. weisen darauf hin, dass unregelmäßige Menstruation und Akne innerhalb von 3 Monaten nach Beginn der Glukokortikoidtherapie rückgängig gemacht werden, während Hirsutismus fast 30 Monate dauert (59). Im Gegensatz zur Kindheit werden in der Adoleszenz häufig länger wirkende Steroide verwendet und Regimes von 5 mg Prednisolon oder 0,25 mg Dexamethason empfohlen (53). In der realen Welt haben klinische Daten jedoch eine Vielzahl unterschiedlicher Therapien gezeigt, die im NCCAH-Management angewendet werden (41).
Das primäre Ziel der Behandlung sollte die Linderung der hyperandrogenen Symptome des Patienten sein. Daher sollten bei Personen mit NCCAH, die ausschließlich aufgrund von Laborergebnissen diagnostiziert wurden, klinische Merkmale die Managementempfehlungen leiten, da hormonelle und molekulare Befunde nicht unbedingt den klinischen Schweregrad vorhersagen. Gemäß den Richtlinien der Endocrine Society sollten NCCAH-Patienten die Möglichkeit erhalten, die GC-Therapie abzubrechen, wenn die Symptome abgeklungen sind (60). Natürlich sollten diese Patienten nicht für die Nachsorge verloren gehen, während die Behandlung im Falle eines erneuten Auftretens der Symptome erneut eingeleitet werden sollte. Darüber hinaus sollten die Patienten im Falle eines Absetzens über die Möglichkeit einer Unfruchtbarkeit informiert und ermutigt werden, ärztlichen Rat einzuholen, wenn sie schwanger werden möchten (19). Es ist zu beachten, dass der entsprechende Transfer des Patienten vom pädiatrischen zum erwachsenen Endokrinologen optimal nach 1 Jahr synchronisierter Überwachung durchgeführt werden sollte (53, 61).
NCCAH und Reproduktion
Subfertilität
Subfertilität wird häufig bei klassischen Formen von CAH berichtet, die hauptsächlich auf Menstruationsstörungen, chronische Anovulation und anatomische Deformitäten zurückzuführen sind. Die Geburtenrate wurde auf 17% im Vergleich zu 65% der Kontrollpopulation geschätzt (62). Inzwischen wurden kürzlich Daten zur Fertilität bei Frauen mit NCCAH im Detail ausgewertet, und die geschätzte Inzidenz von Unfruchtbarkeit beträgt 11% bei NCCAH-Frauen, dh relativ milder als bei CAH, und ist in vielen Fällen leicht zu beheben. Bidet et al. evaluierte die Fruchtbarkeit bei 190 Frauen, die an NCCAH litten, von denen 95 schwanger werden wollten. In dieser Population wurden 187 Schwangerschaften bei 85 Frauen gemeldet, was zu 141 Geburten bei 82 Personen führte. Es muss hervorgehoben werden, dass 99 der Schwangerschaften (52, 9%) vor der Diagnose einer NCCAH auftraten, drei davon unter Anwendung von Ovulationsinduktionsprotokollen, der Rest spontan, während 88 Schwangerschaften nach der NCCAH-Diagnose stattfanden. In der überwiegenden Mehrheit von ihnen entwickelte sich die Schwangerschaft nach der Einführung der Therapie mit Hydrocortison, während es bei 11 Frauen spontan geschah (63, 64). In einem anderen Bericht von 22 beobachteten NCCAH-Frauen, die eine Schwangerschaft wünschten, folgten 12 Schwangerschaften mit Prednison (65).
Fehlgeburten
In der Kohorte von Bidet et al., die Fehlgeburtenrate war 6.5 und 26.3% in Patienten, die mit GCs und unbehandelten Patienten beziehungsweise behandelt sind (64). Der Ausgang der Schwangerschaft kann sogar ohne Glukokortikoidbehandlung in Fällen erfolgreich sein, in denen NCCAH noch nicht diagnostiziert wurde, wie auch in einem Fallbericht von Cuhaci et al. Dennoch erlebte dasselbe Paar zwei Fehlgeburten und berichtete von Subfertilität nach dieser ersten Schwangerschaft (66). Was die Möglichkeit von Frühschwangerschaften betrifft, wie von Bidet et al. es unterscheidet sich nicht wesentlich von dem der übrigen Bevölkerung (15).
Fruchtbarkeitsplanung
Für Frauen mit symptomatischem Hyperandrogenismus oder mit gemeldeter Unfruchtbarkeit, die jedoch schwanger werden möchten, wird eine GC-Therapie dringend empfohlen. Während dieser Zeit sind die verwendeten Glukokortikoide Prednison oder Hydrocortison. Die wichtigste Maßnahme, weil sie die nachfolgenden Schritte leiten wird, ist die genetische Untersuchung der zukünftigen Eltern. Frauen mit NCCAH, die schwanger werden möchten, sollten sich des Risikos bewusst sein, ein Kind mit der klassischen Form der Krankheit zur Welt zu bringen. Das Ergebnis von Schwangerschaften bei Frauen mit NCCAH und insbesondere die Inzidenz von Säuglingen, die mit CCAH geboren wurden, wird in neueren Studien auf 1,5 bis 2,5% geschätzt, wobei NCCAH bei etwa 15% liegt (36, 64). Natürlich hängt diese Häufigkeit von der Referenzpopulation ab, wobei eine höhere Inzidenz in Populationen mit hohen Mischehenraten auftritt (36). Die vorhergesagte Wahrscheinlichkeit, dass Eltern ein Kind mit CAH zur Welt bringen, beträgt 1 zu 120 für den unbekannten väterlichen Genotyp, Null, wenn der Vater keine relevante Mutation aufweist, und 1 zu 4, wenn er eine heterozygote Mutation aufweist (19). Daher ist eine genetische Untersuchung der Partner dieser Frauen unerlässlich, um das Risiko einer Geburt eines Kindes mit der klassischen Form von CAH zu beurteilen (64, 67).
Für den Fall, dass die zukünftige Mutter zwei NC-Mutationen trägt (Tabelle 1), besteht beim zukünftigen Vater keine absolute Notwendigkeit für Gentests, da der Nachwuchs, wenn er für die CYP12A2-Mutationen heterozygot ist, das Risiko hat, in Zukunft die nicht-klassische Form der Krankheit zu entwickeln. Es sollte jedoch beachtet werden, dass der Trägerstatus in verschiedenen Populationen 2-15% beträgt und die Hälfte dieser Individuen eine schwere Mutation trägt. Darüber hinaus sind definitive Empfehlungen für Situationen, in denen keine Gentests erforderlich sind, angesichts der unvollständigen Genotyp-Phänotyp-Korrelation schwierig, insbesondere für mildere Mutationen . Beispielsweise tritt die P30L-Mutation, die am häufigsten mit NCCAH assoziiert ist, auch bei Patienten mit klassischer CAH auf . Eine große multizentrische europäische Studie hat kürzlich gezeigt, dass dies bei 78% der Patienten der Fall war . In dieser Studie war die milde V218L-Mutation in 30% der Fälle eher mit klassischem als mit NCCAH assoziiert. Daher sollten allen Patienten mit NCCAH genetische Beratung und molekulare Beurteilung der Fortpflanzungspartner angeboten werden.
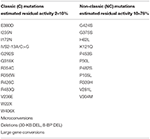
Tabelle 1. Mutationen von CYP21A2 mit minimaler (C) oder moderater (NC) Restenzymaktivität (14, 68, 69).
Im Gegensatz dazu ist für den Fall, dass die zukünftige Mutter eine zusammengesetzte Heterozygote mit einer schweren (C) und einer NC-Mutation ist, eine genetische Untersuchung des zukünftigen Vaters obligatorisch. Wir sollten bedenken, dass Daten, die aus Studien mit Immunoassays oder den genaueren LC-MS / MS-Verfahren stammen, durchweg die Unfähigkeit hervorheben, Heterozygotie unter Verwendung von Basal- oder ACTH-stimulierten 17-OHP-Werten zuverlässig auszuschließen, aufgrund der signifikanten Überlappung mit dem Normalbereich. Man kann die Verwendung eines Synacthen-Tests vorschlagen, und wenn er nicht mit der Heterozygotie kompatibel ist (Summe der Basal- und Peak-stimulierten 17-OHP-Werte < 1,5 nmol / L), könnten DNA-Tests vermieden werden. Wir sollten jedoch bei der Interpretation dieses Tests sehr vorsichtig sein, da er in anderen Studien nicht validiert wurde. Daher kann die Verwendung von ACTH-21-Desoxycortisol entweder einzeln oder als Teil eines Steroidverhältnisses oder Steroidprofils die biochemische Identifizierung von Heterozygoten in der Zukunft erleichtern, insbesondere wenn LC-MS / MS breiter verfügbar wird . Wenn der zukünftige Vater eine NC-Mutation trägt, ist nichts anderes erforderlich. Umgekehrt, wenn er Träger einer klassischen Mutation ist, stellt sich die Frage nach der pränatalen Behandlung des Fötus. Alle möglichen Genotypkombinationen und die vorgeschlagenen Verfahren sind in Tabelle 2 dargestellt.

Tabelle 2. Planung der vorgeschlagenen Verfahren während der Schwangerschaft basierend auf dem Genotyp der zukünftigen Eltern.
Pränatale Behandlung von CAH
Das Ziel der pränatalen Behandlung von CAH ist die Verhinderung der genitalen Virilisierung des Fötus, aber auch die Linderung des Stresses der Eltern, die wahrscheinlich ein Kind mit mehrdeutigen Genitalien haben (70). Dexamethason wird wegen seiner Fähigkeit verwendet, die Plazenta zu überqueren und weil es nicht von der plazentaren 11β OH-Steroid-Dehydrogenase deaktiviert wird und nur minimal an das Cortisol-bindende Globulin (CBG) der Mutter im Blut bindet. Dexamethason verringert durch Unterdrückung der fetalen ACTH-Sekretion die erhöhte fetale Androgenproduktion. Die Behandlung muss abgebrochen werden, falls die Karyotyp- oder DNA-Analyse ein Männchen bzw. ein nicht betroffenes Weibchen ergibt (71). Obwohl die fetale Genitalvirilisierung bereits 6-7 Wochen nach der Empfängnis begonnen hat, können Chorionzottenbiopsien zur genetischen Diagnose nicht vor der 10. bis 12. Dieses Zeitintervall legt nahe, dass alle Schwangerschaften mit einem Risiko für virilisierende CAH behandelt werden sollten, obwohl nur 1 von 8 Feten betroffen und weiblich ist (19, 34, 70).
Ob diese Exposition des Fötus gegenüber Dexamethason für vorbeugende Maßnahmen medizinisch und ethisch akzeptabel ist, bleibt umstritten (70). Tierstudien haben gezeigt, dass Dexamethason im ersten Trimester das Geburtsgewicht senkt, die Betazellfunktion der Nieren und die Gehirnentwicklung beeinflusst und im Erwachsenenalter zu Angstzuständen, Bluthochdruck und Hyperglykämie prädisponiert (70). Darüber hinaus klagen 10-20% der schwangeren Frauen, die Dexamethason während der Schwangerschaft einnehmen, über Gewichtszunahme, gesteigerten Appetit, Stimmungsschwankungen, Schlaflosigkeit, Ödeme, Bluthochdruck, Hyperglykämie und Striae. New et al. fand einen unterdurchschnittlichen Prader-Score bei Feten, die pränatal mit Dexamethason behandelt wurden, aber keinen Unterschied in den Langzeitergebnissen (72). Zusätzlich wurde in einem Review von Merce Fernandez-Balsells et al. Es wurde gezeigt, dass Dexamethason mit einer Verringerung der fetalen Virilisierung ohne signifikante nachteilige Auswirkungen auf die Mutter oder den Fötus verbunden ist (63). Die Autoren der oben genannten Übersicht sowie die konsekutiven Richtlinien der Endocrine Society weisen jedoch darauf hin, dass das Thema aufgrund der geringen Stichprobengröße im gesamten Literaturbestand ungewiss bleibt und weitere Untersuchungen eindeutig erforderlich sind.
Die Entscheidung über die Einleitung der Behandlung sollte nur in großen Zentren mit einem erfahrenen Team und Protokollen getroffen werden, die von institutionellen Prüfungsausschüssen genehmigt wurden und auf den Werten und Vorlieben der Familie und mit deren schriftlicher Einverständniserklärung als Voraussetzung beruhen (19, 63). Mehrere Spezialisten auf diesem Gebiet weisen darauf hin, dass ein höherer Wert darauf gelegt werden sollte, eine unnötige pränatale Exposition von Mutter und Fötus gegenüber Dexamethason zu verhindern, als Eltern und Patienten den emotionalen Tribut mehrdeutiger Genitalien aufzuerlegen. Vor allem sollte den Eltern klargestellt werden, dass die Verabreichung von Dexamethason den Patientenstatus nicht verändert, sondern darauf abzielt, die Notwendigkeit einer Operation zu verringern, anstatt das Leben oder die intellektuellen Fähigkeiten zu erhalten. Sie müssen auch wissen, dass die Wahrscheinlichkeit, dass ihr Kind an der klassischen Form der Krankheit leidet, trotz Behandlung hoch ist. Am wichtigsten ist jedoch, dass die Exposition eines jungen Organismus gegenüber einem sehr potenten GC während einer besonders sensiblen Phase der fetalen Programmierung und des Wachstums, die sich bei einem Fötus mit dem XY-Karyotyp als nutzlos erweisen könnte, derzeit nicht durch robuste und unbestreitbare Daten gestützt wird.
Idealerweise sollte eine frühzeitige Diagnose erfolgen, die über eine nicht-invasive Technik und vor Beginn der genitalen Virilisierung des Fötus erfolgt. In dieser Richtung weisen neuartige Studien auf die Verwendung von zellfreier fetaler DNA aus mütterlichem Plasma als vielversprechende Methode hin, die die Bestimmung des fetalen Geschlechts und die Diagnose von CAH in einem frühen Gestationsalter (<9 Wochen). Wenn dieses Verfahren in der klinischen Praxis weit verbreitet ist, wird eine unnötige pränatale Dexamethasonbehandlung vermieden (73).
Behandlung während der Schwangerschaft
Während der Schwangerschaft sollten Frauen mit NCCAH vorzugsweise mit Hydrocortison behandelt werden, nicht nur, weil die Schwangerschaft typischerweise mit einem erhöhten Stresslevel einhergeht, sondern auch als vorbeugende Maßnahme gegen die oben erwähnte hohe Inzidenz von Fehlgeburten. Diese Form von Glucocorticoid wird aufgrund seiner Metabolisierung durch das Plazentaenzym 11β OH-Steroiddehydrogenase II begünstigt, wodurch verhindert wird, dass Glucocorticoid eine Wirkung auf den Fötus hat (71). Zum Zeitpunkt der Empfängnis sollte die Hydrocortisondosis auf 20-25 mg / Tag erhöht werden, und alle 6-8 Wochen werden Dosisanpassungen durchgeführt, um den Testosteronspiegel auf dem oberen Normalniveau des Schwangerschaftstrimesters zu halten (74). Als Beispiel werden die Testosteron- und D4-Werte eines Patienten mit NCCAH von der Empfängnis bis zu 3 Jahren nach der Entbindung von zwei gesunden Zwillingen gezeigt (Abbildung 1).
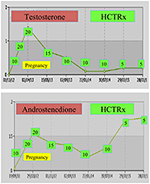
Abbildung 1. Testosteron- und Andostendion-Spiegel und anschließende Hydrocortison-Dosierung (HCTRx) von der Empfängnis bis 3 Jahre nach der Entbindung an einen Patienten mit NCCAH verabreicht.
Weitere Sonderthemen
Stressmanagement bei NCCAH
Bei schwerem lebensbedrohlichen Stress, Operationen oder schweren Erkrankungen können Patienten mit NCCAH, die mit Glukokortikoiden behandelt werden, größere oder häufigere Dosen von Glukokortikoiden benötigen, da ihre Nebennierenfunktion iatrogen unterdrückt ist. Es ist daher von entscheidender Bedeutung, die Eltern kleiner Kinder sowie die Patienten über ihren Übergang zur Erwachsenenpflege über die Stressdosierung aufzuklären. Bei NCCAH-Patienten, die nicht mit GCs behandelt werden, oder bei Absetzen sollte das Cortisol-Ansprechen auf ACTH beurteilt werden. Fast ein Drittel der NCCAH-Patienten spricht unzureichend auf die Stimulation an (15, 75). Für diejenigen, die normal auf die ACTH-Stimulation ansprechen, wird keine Behandlung mit Stressdosen empfohlen (19). Eine Mineralocorticoid-Therapie ist in keinem der Fälle erforderlich, da diese Patienten eine normale Aldosteronsekretion haben und keine Salzverschwendung entwickeln (58). Bemerkenswert ist, dass im Falle einer Hyperthyreose oder einer Erhöhung der Levothyroxin-Behandlung die Cortisol-Clearance erhöht ist und eine Nebennierenkrise auftreten kann (58, 76).
Psychobiologische Aspekte im NCCAH
Meyer-Bahlburg et al. haben in mehreren Studien berichtet beeinträchtigtes psychologisches Profil bei Patienten mit NCCAH aufgrund von 21OH-Mangel. Insbesondere hatten die betroffenen Frauen einen höheren Maskulinisierungs- / Defeminisierungs-Score bei mehreren Maßnahmen des geschlechtsbezogenen Verhaltens im Vergleich zu normalen Kontrollfrauen, wenn auch deutlich weniger als bei Frauen mit klassischer CAH. Sie hatten auch eine signifikant erhöhte bisexuelle oder homosexuelle Orientierung. Retrospektive klinisch-qualitative Interviews mit diesen Frauen zeigten eine Vorgeschichte von Beschwerden und sozialem Stress im Zusammenhang mit ihren Erfahrungen vor der Behandlung mit androgenabhängigen Anzeichen wie Akne, Hirsutismus und Empfängnisschwierigkeiten (59, 77, 78). In ähnlicher Weise Arlt et al. zeigte, dass der subjektive Gesundheitszustand in allen acht Bereichen einer kurzfristigen Gesundheitsumfrage signifikant beeinträchtigt war, wobei die auffälligsten Unterschiede im Vergleich zu alters- und geschlechtsangepassten Kontrollen in Bezug auf die Bereiche allgemeine Gesundheit, Vitalität und Rollenbeschränkungen aufgrund emotionaler Probleme auftraten. Der Fragebogen der Hospital Anxiety and Depression Scale (HADS) ergab ebenfalls erhöhte Angstwerte (41). Unter Berücksichtigung dieser Befunde sollten psychologische Parameter zur Therapieleitung bei Frauen mit NCCAH berücksichtigt werden, und im Rahmen des patientenorientierten Ansatzes muss eine psychologische Diagnose und Unterstützung angeboten werden.
Schlussfolgerungen
NCCAH wird aufgrund der Beibehaltung der Enzymaktivität von 20-50% als die häufigere und mildere Form von CAH angesehen. Die meisten Patienten können in jedem Stadium ihres Lebens aufgrund von Symptomen im Zusammenhang mit einem Androgenüberschuss ärztlichen Rat einholen. Dazu gehören vorzeitige Pubarche, schnelles Wachstum, Hirsutismus, Akne, Menstruationsstörungen oder Unfruchtbarkeit sowie viele andere weniger prominente Manifestationen. Frühmorgendliche Ausgangswerte von 17 OHP als guter erster Screening-Test und weitere Bewertung mit ACTH-Stimulation und, im Falle von Borderline-Ergebnissen, Gentests, wird empfohlen. In der Regel sollten Androstendion- und nicht 17 OHP-Spiegel während der Nachsorge verwendet werden.
Die Behandlungsentscheidung sollte auf der Beurteilung der Fakten beruhen und einem individuellen Ansatz folgen. Die Behandlung ist nicht immer angezeigt, es sei denn, der Patient ist symptomatisch, z. B. Kinder mit früh einsetzendem und schnellem Fortschreiten der Scham- und Körperbehaarung, schnellem Wachstum und / oder Skelettfortschritt oder Frauen mit Oligomenorrhoe, Akne, Hirsutismus, Unfruchtbarkeit oder einer Kombination dieser und anderer der oben genannten Symptome. Genetische Beratung wird bei NCCAH-Frauen, die schwanger werden möchten, sowie die Genotypisierung des Vaters dringend empfohlen. Das Gesamtmanagement des Patienten umfasst zusätzlich das Management der wahrscheinlichen Komplikationen der Glukokortikoidtherapie oder stoffwechselbedingter Manifestationen der Krankheit. Da viele therapeutische Fragen im Zusammenhang mit der angemessenen Behandlung dieser Patienten noch nicht geklärt sind, ist es für den behandelnden Arzt sehr wichtig, über alle Entwicklungen auf diesem Gebiet auf dem Laufenden zu bleiben und die neuen Daten in seine klinische Praxis zu integrieren. Sicherlich sind weitere klinische Studien in diesem Bereich unerlässlich.Zusammenfassend lässt sich sagen, dass für die NCCAH-Frau der ideale Ansatz ein maßgeschneiderter Ansatz ist, der einen reibungslosen Übergang ihres Managements beinhaltet, sobald sie vom pädiatrischen zum erwachsenen Endokrinologen überwiesen wird, zusammen mit einer symptomorientierten Behandlung, die sie ihr ganzes Leben lang begleiten wird. Angesichts der zahlreichen Faktoren, die das hyperandrogene System beeinflussen, ist es ratsam, die Patienten zu einem gesünderen Lebensstil zu ermutigen, indem sie ihre Ernährungsgewohnheiten verbessern, die Bewegung steigern und eine Gewichtsreduktion anstreben. Darüber hinaus ist in bestimmten Fällen psychologische Unterstützung oft von Vorteil. Darüber hinaus benötigen Spezialisten in Bereichen, die an der Behandlung dieser Störung beteiligt sind, wie Dermatologen, Gynäkologen und Psychologen, ein tiefes Verständnis für das Management verdächtiger oder bereits diagnostizierter Fälle von NCCAH. Denken wir abschließend an das aufschlussreiche Zitat der amerikanischen Astronomin Vera Cooper Rubin: „Die Wissenschaft schreitet am besten voran, wenn Beobachtungen uns zwingen, unsere Vorurteile zu ändern.“
Autorenbeiträge
Alle aufgeführten Autoren haben einen wesentlichen, direkten und intellektuellen Beitrag zum Werk geleistet und es zur Veröffentlichung freigegeben.
Erklärung zum Interessenkonflikt
Die Autoren erklären, dass die Forschung in Abwesenheit von kommerziellen oder finanziellen Beziehungen durchgeführt wurde, die als potenzieller Interessenkonflikt ausgelegt werden könnten.
1. Speiser PW, Weiß Stk. Angeborene Nebennierenhyperplasie. In: N Engl J Med. (2003) 349:776–88. doi: 10.1056/NEJMra021561
PubMed Zusammenfassung | CrossRef Volltext / Google Scholar
2. Turcu AF, Auchus RJ. Adrenal steroidogenesis and congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. (2015) 44:275–96. doi: 10.1016/j.ecl.2015.02.002
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Speiser PW. Nonclassic adrenal hyperplasia. Rev Endocr Metab Disord. (2009) 10:77–82. doi: 10.1007/s11154-008-9097-x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Gidlöf S, Falhammar H, Thilén A, von Döbeln U, Ritzén M, Wedell A, et al. Einhundert Jahre angeborene Nebennierenhyperplasie in Schweden: eine retrospektive, bevölkerungsbasierte Kohortenstudie. In: Lancet Diabetes Endocrinol. (2013) 1:35–42. doi: 10.1016/S2213-8587(13)70007-X
PubMed Zusammenfassung | CrossRef Volltext | Google Scholar
5. Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, Neu MI. Hohe Häufigkeit von nicht-klassischem Steroid-21-Hydroxylase-Mangel. Bin J Hum Genet. (1985) 37:650–67.
PubMed Abstract | Google Scholar
6. Wilson RC, Nimkarn S, Dumic M, Obeid J, Azar MR, Azar M, et al. Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Mol Genet Metab. (2007) 90:414–21. doi: 10.1016/j.ymgme.2006.12.005
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Miller WL. Clinical review 54: genetics, diagnosis, and management of 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1994) 78:241–6. doi: 10.1210/jcem.78.2.8106606
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Witchel SF, Azziz R. Nonclassic congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2010) 2010:625105. doi: 10.1186/1687-9856-2010-625105
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Wilson RC, Mercado AB, Cheng KC, Neu MI. Steroid-21-Hydroxylase-Mangel: Genotyp kann Phänotyp nicht vorhersagen. In: J Clin Endocrinol Metab. (1995) 80:2322–9. Ursprungsbezeichnung: 10.1210/jc.80.8.2322
CrossRef Volltext / Google Scholar
10. Weiß PC. Neugeborenen-Screening auf angeborene Nebennierenhyperplasie. Nat Rev Endocrinol. (2009) 5:490–8. doi: 10.1038/nr.2009.148
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Kohn B, Levine LS, Pollack MS, Pang S, Lorenzen F, Levy D, et al. Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. (1982) 55:817–27. doi: 10.1210/jcem-55-5-817
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Moran C, Azziz R, Carmina E, Dewailly D, Fruzzetti F, Ibañez L, et al. 21-Hydroxylase-defiziente nichtklassische Nebennierenhyperplasie ist eine progressive Erkrankung: eine multizentrische Studie. Bin J Geburtshelfer Gynecol. (2000) 183:1468–74. doi: 10.1067 / mob.2000.108020
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
13. Azziz R, Hincapie LA, Knochenhauer ES, Dewailly D, Fuchs L, Stiefel LR. Screening auf 21-Hydroxylase-defiziente nichtklassische Nebennierenhyperplasie bei hyperandrogenen Frauen: eine prospektive Studie. Fertil Steril. (1999) 72:915–25. doi: 10.1016/S0015-0282(99)00383-0
PubMed Zusammenfassung | CrossRef Volltext | Google Scholar
14. Es sind keine frei zugänglichen ergänzenden Materialien verfügbar in: Livadas S, Dracopoulou M, Dastamani A, Sertedaki A, Maniati-Christidi M, Magiakou A-M, et al. Das Spektrum der klinischen, hormonellen und molekularen Befunde bei 280 Personen mit nichtklassischer kongenitaler Nebennierenhyperplasie, die durch Mutationen des CYP21A2-Gens verursacht wurde. In: Clin Endocrinol (Oxf). (2015) 82:543–9. doi: 10.1111/cen.12543
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
15. Bidet M, Bellanné-Chantelot C, Galand-Portier M-B, Tardy V, Billaud L, Laborde K, et al. Klinische und molekulare Charakterisierung einer Kohorte von 161 nicht verwandten Frauen mit nichtklassischer kongenitaler Nebennierenhyperplasie aufgrund eines 21-Hydroxylase-Mangels und 330 Familienmitgliedern. In: J Clin Endocrinol Metab. (2009) 94:1570–8. Ursprungsbezeichnung: 10.1210/jc.2008-1582
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
16. Escobar-Morreale HF, Sanchón R, San Millán JL. Eine prospektive Studie zur Prävalenz der nichtklassischen kongenitalen Nebennierenhyperplasie bei Frauen mit hyperandrogenen Symptomen und Anzeichen. In: J Clin Endocrinol Metab. (2008) 93:527–33. Ursprungsbezeichnung: 10.1210/jc.2007-2053
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
17. Dewailly D, Vantyghem-Haudiquet MC, Sainsard C, Buvat J, Cappoen JP, Ardaens K, et al. Klinische und biologische Phänotypen bei spät einsetzendem 21-Hydroxylase-Mangel. In: J Clin Endocrinol Metab. (1986) 63:418–23. doi: 10.1210/jcem-63-2-418
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
18. Gönç EN, Ozön ZA, Alikaşifoglu A, Engiz O, Bulum B, Kandemir N. (2011). Ist Basalserum-17-OH-Progesteron ein zuverlässiger Parameter zur Vorhersage der nichtklassischen kongenitalen Nebennierenhyperplasie bei vorzeitiger Adrenarche? Türke Jr. 53:274–80.
PubMed Zusammenfassung / Google Scholar
19. Es sind keine frei zugänglichen ergänzenden Materialien verfügbar Zitationen Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Merke DP, et al. Angeborene Nebennierenhyperplasie aufgrund eines Steroid-21-Hydroxylase-Mangels: eine klinische Praxisrichtlinie der Endocrine Society. In: J Clin Endocrinol Metab. (2010) 95:4133–60. Ursprungsbezeichnung: 10.1210 / jc.2009-2631
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Dacou-Voutetakis C, Dracopoulou M. High incidence of molecular defects of the CYP21 gene in patients with premature adrenarche. J Clin Endocrinol Metab. (1999) 84:1570–4. doi: 10.1210/jc.84.5.1570
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Ambroziak Y, Kepczynska-Events Und, Kurylović, Małunowicz EM, Wójcicka, Miskevic P, et al. The diagnosis of nonclassic angeborenen nebennierenhyperplasie due to 21-hydroxylase deficiency, based on basal Serum or post-ACTH stimulation 17-hydroxyprogesterone, can lead to false-positive diagnosis. Clin Endocrinol (Oxf). (2016) 84:23–9. doi: 10.1111 / Preis.12935
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Ambroziak Haben, Kepczynska-Ereignisse Und Курылович, Wysłouch-Cieszyńska A, Małunowicz EM, Bartoszewicz, et al. LC-MS/MS improves screening towards 21-hydroxylase deficiency. Gynecol ist Endokrinol. (2015) 31:296–300. doi: 10.3109/09513590.2014.994599
PubMed Abstract | CrossRef Full Text | Google Scholar
23. BBC, CC, Goldman MM, Zhang K, Clark-N, Reitz RE Welt CK. Gleichzeitige Messung von dreizehn Steroidhormonen bei Frauen mit polyzystischem Ovarialsyndrom und Kontrollfrauen mittels Flüssigkeitschromatographie-Tandem-Massenspektrometrie. Plus Eins. (2014) 9:e93805. doi: 10.1371/Zeitschrift.pone.0093805
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
24. Ray JA, Kushnir MM, Yost RA, Rockwood AL, Wayne Meikle A. Leistungssteigerung bei der Messung von 5 endogenen Steroiden durch LC-MS / MS kombiniert mit differentieller Ionenmobilitätsspektrometrie. Clin Chim Acta. (2015) 438:330–6. ursprungsbezeichnung: 10.1016 / j.cca.2014.07.036
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
25. Stoupa A, González-Briceño L, Pinto G, Samara-Boustani D, Thalassinos C, Flechtner I, et al. Unzureichende Cortisolantwort auf den Tetracosactid (Synacthen®) -Test bei nicht-klassischer kongenitaler Nebennierenhyperplasie: eine Ausnahme von der Regel? In: Horm Res Paediatr. (2015) 83:262–7. doi: 10.1159/000369901
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. (1992) 90:584–95. doi: 10.1172/JCI115897
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Temeck JW, Pang SY, Nelson C, Neu MI. Genetische Defekte der Steroidogenese bei vorzeitiger Pubarche. In: J Clin Endocrinol Metab. (1987) 64:609–17. doi: 10.1210/jcem-64-3-609
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
28. Voutilainen R, Jääskeläinen J. Vorzeitige Adrenarche: Ätiologie, klinische Befunde und Konsequenzen. In: J Steroid Biochem Mol Biol. (2015) 145:226–36. doi: 10.1016/j.jsbmb.2014.06.004
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Oberfield SE, Sopher AB, Gerken AT. Approach to the girl with early onset of pubic hair. J Clin Endocrinol Metab. (2011) 96:1610–22. doi: 10.1210/jc.2011-0225
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Janus D, Wojcik M, Malunowicz E, Starzyk JB. Ein Fall von rezidivierenden labialen Adhäsionen bei einem 15 Monate alten Kind mit asymptomatischer nicht-klassischer kongenitaler Nebennierenhyperplasie aufgrund eines 21-Hydroxylase-Mangels. In: J Pediatr Endocrinol Metab. (2012) 25:1017–21. doi: 10.1515/jpem-2012-0190
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
31. Gopal-Kothandapani JS, Petkar A, O’Shea E, Banerjee I. Perianales Haar als ungewöhnliche Darstellung der nicht-klassischen kongenitalen Nebennierenhyperplasie. BMJ Fall Rep. (2013) 2013: bcr2013010123. doi: 10.1136/bcr-2013-010123
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Falhammar H, Nordenström A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine. (2015) 50:32–50. doi: 10.1007/s12020-015-0656-0
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Chrousos GP, Loriaux DL, Mann DL, Cutler GB. Late-onset 21-hydroxylase deficiency mimicking idiopathic hirsutism or polycystic ovarian disease. Ann Intern Med. (1982) 96:143–8. doi: 10.7326/0003-4819-96-2-143
CrossRef Full Text | Google Scholar
34. Witchel SF. Non-classic congenital adrenal hyperplasia. Steroids. (2013) 78:747–50. doi: 10.1016/j.steroids.2013.04.010
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Carmina E, Rosato F, Jannì A, Rizzo M, Longo RA. Extensive clinical experience: relative prevalence of different androgen excess disorders in 950 women referred because of clinical hyperandrogenism. J Clin Endocrinol Metab. (2006) 91:2–6. doi: 10.1210/jc.2005-1457
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Moran C, Azziz R, Weintrob N, Witchel SF, Rohmer V, Dewailly D, et al. Reproduktives Ergebnis von Frauen mit 21-Hydroxylase-defizienter nichtklassischer Nebennierenhyperplasie. In: J Clin Endocrinol Metab. (2006) 91:3451–6. Ursprungsbezeichnung: 10.1210/jc.2006-0062
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
37. Levin JH, Carmina E, Lobo RA. Ist die unangemessene Gonadotropinsekretion von Patienten mit polyzystischem Ovarialsyndrom der von Patienten mit angeborener Nebennierenhyperplasie im Erwachsenenalter ähnlich? Fertil Steril. (1991) 56:635–40. doi: 10.1016/S0015-0282(16)54592-0
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Carmina E, Lobo RA. Ovarian suppression reduces clinical and endocrine expression of late-onset congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Fertil Steril. (1994) 62:738–43. doi: 10.1016/S0015-0282(16)56998-2
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Ludwig E. Klassifikation der Arten der androgenetischen Alopezie (häufige Kahlheit) beim weiblichen Geschlecht. In: Br J Dermatol. (1977) 97:247–54. doi: 10.1111/j.1365-2133.1977.tb15179.x
PubMed Zusammenfassung | CrossRef Volltext | Google Scholar
40. Trakakis E, Papadavid E, Dalamaga M, Koumaki D, Stavrianeas N, Rigopoulos D, et al. Prävalenz der nicht-klassischen kongenitalen Nebennierenhyperplasie aufgrund von 21-Hydroxylase-Mangel bei griechischen Frauen mit Akne: eine Krankenhaus-basierte Querschnittsstudie. In: J Eur Acad Dermatol Venereol. (2013) 27:1448–51. Ursprungsbezeichnung: 10.1111/j.1468-3083.2012.04613.x
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
41. Arlt W, Willis DS, Wild SH, Krone N, Doherty EJ, Hahner S, et al. Gesundheitszustand von Erwachsenen mit kongenitaler Nebennierenhyperplasie: eine Kohortenstudie mit 203 Patienten. In: J Clin Endocrinol Metab. (2010) 95:5110–21. Ursprungsbezeichnung: 10.1210/jc.2010-0917
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
42. Saygili F, Oge A, Yilmaz C. Hyperinsulinämie und Insulinunempfindlichkeit bei Frauen mit nichtklassischer kongenitaler Nebennierenhyperplasie aufgrund eines 21-Hydroxylase-Mangels: die Beziehung zwischen Serumleptinspiegeln und chronischer Hyperinsulinämie. Horm Res. (2005) 63:270-4. doi: 10.1159/000086363
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Speiser PW, Serrat J, New MI, Gertner JM. Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1992) 75:1421–4. doi: 10.1210/jc.75.6.1421
PubMed Abstract | CrossRef Full Text | Google Scholar
44. Han TS, Stimson RH, Rees DA, Krone N, Willis DS, Conway GS, et al. Glukokortikoid-Behandlungsschema und Gesundheitsergebnisse bei Erwachsenen mit angeborener Nebennierenhyperplasie. In: Clin Endocrinol. (2013) 78:197–203. doi: 10.1111/cen.12045
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
45. Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Verwendung von oralen Kortikosteroiden und Frakturrisiko. J Bone Miner Res. (2000) 15:993-1000. Ursprungsbezeichnung: 10.1359/jbmr.2000.15.6.993
PubMed Abstract | CrossRef Full Text | Google Scholar
46. Nebesio TD, Renbarger JL, Nabhan ZM, Ross SE, Slaven JE, Li L, et al. Differential effects of hydrocortisone, prednisone, and dexamethasone on hormonal and pharmacokinetic profiles: a pilot study in children with congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2016) 2016:17. doi: 10.1186/s13633-016-0035-5
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
47. Weintrob N, Dickerman Z, Sprecher E, Galatzer A, Pertzelan A. Nicht-klassischer 21-Hydroxylase-Mangel im Säuglings- und Kindesalter: Der Einfluss des Therapiebeginns auf die Pubertät und die Endgröße. In: Eur J Endocrinol. (1997) 136:188–95. doi: 10.1530/eje.0.1360188
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Bonfig W, Bechtold S, Schmidt H, Knorr D, Schwarz HP. Reduced final height outcome in congenital adrenal hyperplasia under prednisone treatment: deceleration of growth velocity during puberty. J Clin Endocrinol Metab. (2007) 92:1635–9. doi: 10.1210/jc.2006-2109
PubMed Abstract | CrossRef Full Text | Google Scholar
49. Dörr HG. Wachstum bei Patienten mit klassischer kongenitaler Nebennierenhyperplasie aufgrund eines 21-Hydroxylase-Mangels. Horm Res. (2007) 68(Ergänzung 5): 93-9. doi: 10.1159/000110587
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
50. Hoepffner W, Kaufhold A, Willgerodt H, Keller E. Patienten mit klassischer kongenitaler Nebennierenhyperplasie aufgrund eines 21-Hydroxylase-Mangels können ihre Zielgröße erreichen: die Leipziger Erfahrung. Horm Res. (2008) 70:42-50. doi: 10.1159/000129677
PubMed Abstract | CrossRef Full Text | Google Scholar
51. van der Kaay D, van den Akker E. Ultralow-dose dexamethasone to preserve endogenous cortisol stress response in nonclassical congenital adrenal hyperplasia: a new promising treatment. Int J Endocrinol Metab. (2014) 12:e14657. doi: 10.5812/ijem.14657
CrossRef Full Text | Google Scholar
52. Lin-Su K, Vogiatzi MG, Marshall I, Harbison MD, Macapagal MC, Betensky B, et al. Treatment with growth hormone and luteinizing hormone releasing hormone analog improves final adult height in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. (2005) 90:3318–25. doi: 10.1210/jc.2004-2128
PubMed Abstract | CrossRef Full Text | Google Scholar
53. McCann-Crosby B, Chen M-J, Lyons SK, Lin Y, Axelrad M, Dietrich JE, et al. Nonclassical congenital adrenal hyperplasia: ziele der Behandlung und des Übergangs. Pediatr Endocrinol Rev. (2014) 12:224-38.
PubMed Zusammenfassung / Google Scholar
54. Ogilvie CM, Hocke NS, Rumsby G, Creighton SM, Liao L-M, Conway GS. Angeborene Nebennierenhyperplasie bei Erwachsenen: eine Überprüfung der medizinischen, chirurgischen und psychologischen Fragen. In: Clin Endocrinol. (2006) 64:2–11. doi: 10.1111/j.1365-2265.2005.02410.x
PubMed Abstract | CrossRef Full Text | Google Scholar
55. Mah PM, Jenkins RC, Rostami-Hodjegan A, Newell-Price J, Doane A, Ibbotson V, et al. Weight-related dosing, timing and monitoring hydrocortisone replacement therapy in patients with adrenal insufficiency. Clin Endocrinol. (2004) 61:367–75. doi: 10.1111/j.1365-2265.2004.02106.x
PubMed Abstract | CrossRef Full Text | Google Scholar
56. Diaz A, Laufer HERR Breech LLAA von PC auf A, Pflege AC von O und GC auf AH. Menstruation bei Mädchen und Jugendlichen: Verwendung des Menstruationszyklus als Vitalzeichen. Diatrie. (2006) 118:2245–50. doi: 10.1542/peds.2006-2481
Querverweis Volltext / Google Scholar
57. Merke DP, Poppas DP. Management von Jugendlichen mit angeborener Nebennierenhyperplasie. in: lancet Diabetes Endocrinol. (2013) 1:341–52. doi: 10.1016/S2213-8587(13)70138-4
PubMed Abstract | CrossRef Full Text | Google Scholar
58. Verma S, Green-Golan L, VanRyzin C, Drinkard B, Mehta SP, Weise M, et al. Adrenomedullary function in patients with nonclassic congenital adrenal hyperplasia. Horm Metab Res. (2010) 42:607–12. doi: 10.1055/s-0030-1253385
PubMed Abstract | CrossRef Full Text | Google Scholar
59. New MI. Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2006) 91:4205–14. doi: 10.1210/jc.2006-1645
PubMed Abstract | CrossRef Full Text | Google Scholar
60. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2018) 103:4043–88. doi: 10.1210/jc.2018-01865
PubMed Abstract | CrossRef Full Text | Google Scholar
61. Kruse B, Riepe FG, Krone N, Bosinski HAG, Kloehn S, Partsch CJ, et al. Congenital adrenal hyperplasia – how to improve the transition from adolescence to adult life. Exp Clin Endocrinol Diabetes. (2004) 112:343–55. doi: 10.1055/s-2004-821013
PubMed Abstract | CrossRef Full Text | Google Scholar
62. Es sind keine frei zugänglichen ergänzenden Materialien verfügbar Zitationen Gastaud F, Brauner R, Duranteau L, Kutten F, et al. Beeinträchtigte sexuelle und reproduktive Ergebnisse bei Frauen mit klassischen Formen der kongenitalen Nebennierenhyperplasie. In: J Clin Endocrinol Metab. (2007) 92:1391–6. Ursprungsbezeichnung: 10.1210/jc.2006-1757
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
63. Mercè Fernández-Balsells M, Muthusamy K, Smushkin G, Lampropulos JF, Elamin MB, Abu Elnour NO, et al. Pränatale Dexamethason-Anwendung zur Vorbeugung der Virilisierung bei Schwangerschaften mit einem Risiko für eine klassische kongenitale Nebennierenhyperplasie aufgrund eines 21-Hydroxylase-Mangels (CYP21A2): eine systematische Überprüfung und Metaanalysen. In: Clin Endocrinol. (2010) 73:436–44. Ursprungsbezeichnung: 10.1111/j.1365-2265.2010.03826.x
CrossRef Volltext / Google Scholar
64. Bidet M., Bellanné-Chantelot C., Galand-Portier MB, Golmard JL, Spät V., Morel Y., et al. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2010) 95:1182–90. doi: 10.1210/jc.2009-1383
PubMed Abstract | CrossRef Full Text | Google Scholar
65. Birnbaum MD, Rose LI. Late onset adrenocortical hydroxylase deficiencies associated with menstrual dysfunction. Obstet Gynecol. (1984) 63:445–51.
PubMed Abstract | Google Scholar
66. Cuhaci N, Aydin C, Yesilyurt A, Pinarli FA, Ersoy R, Cakir B. Nonclassical congenital adrenal hyperplasia and pregnancy. Case Rep Endocrinol. (2015) 2015:296924. doi: 10.1155/2015/296924
PubMed Abstract | CrossRef Full Text | Google Scholar
67. Deneux C, Tardy V, Anne D, Mornet E, Billaud L, Charron D, et al. Phänotyp-Genotyp-Korrelation bei 56 Frauen mit nichtklassischer kongenitaler Nebennierenhyperplasie aufgrund eines 21-Hydroxylase-Mangels. In: J Clin Endocrinol Metab. (2001) 86:207–13. doi: 10.1210/jcem.86.1.7131
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
68. Simonetti L, Bruque CD, Fernández CS, Benavides-Mori B, Delea M, Kolomenski JE, et al. CYP21A2-Mutations-Update: Umfassende Analyse von Datenbanken und veröffentlichten genetischen Varianten. Hum Mutat. (2018) 39:5–22. doi: 10.1002/humu.23351
PubMed Abstract | CrossRef Full Text | Google Scholar
69. OMIM Entry – * 613815 – CYTOCHROME P450 FAMILY 21 SUBFAMILY A POLYPEPTIDE 2; CYP21A2.
Google Scholar
70. Miller WL, Witchel SF. Prenatal treatment of congenital adrenal hyperplasia: risks outweigh benefits. Am J Obstet Gynecol. (2013) 208:354–9. doi: 10.1016/j.ajog.2012.10.885
PubMed Abstract | CrossRef Full Text | Google Scholar
71. Lekarev O, New MI. Adrenal disease in pregnancy. Best Pract Res Clin Endocrinol Metab. (2011) 25:959–73. doi: 10.1016/j.beem.2011.08.004
PubMed Abstract | CrossRef Full Text | Google Scholar
72. New MI, Abraham M, Yuen T, Lekarev O. An update on prenatal diagnosis and treatment of congenital adrenal hyperplasia. Semin Reprod Med. (2012) 30:396–9. doi: 10.1055/s-0032-1324723
PubMed Abstract | CrossRef Full Text | Google Scholar
73. Kazmi D, Bailey J, Yau M, Abu-Amer W, Kumar A, Low M, et al. New developments in prenatal diagnosis of congenital adrenal hyperplasia. J Steroid Biochem Mol Biol. (2017) 165:121–3. doi: 10.1016/j.jsbmb.2016.06.016
PubMed Abstract | CrossRef Full Text | Google Scholar
74. Ritzén EM, Oberfield SE, Sippell WG, Speiser PW, et al. Konsenserklärung zum 21-Hydroxylase-Mangel der Europäischen Gesellschaft für Pädiatrische Endokrinologie und der Lawson wilkins pediatric endocrine society. Horm Res. (2002) 58:188-95. doi: 10.1159/000065490
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar
75. Nandagopal R, Sinaii N, Avila NA, Van Ryzin C, Chen W, Finkielstain GP, et al. Phänotypische Profilerstellung von Eltern mit kryptischer nichtklassischer kongenitaler Nebennierenhyperplasie: Befunde in 145 nicht verwandten Familien. In: Eur J Endocrinol. (2011) 164:977–84. doi: 10.1530/EJE-11-0019
PubMed Zusammenfassung | CrossRef Volltext | Google Scholar
76. Takasu N, Nakachi K, Higa H. Die Entwicklung der Basedow-Hyperthyreose verursachte eine Nebennierenkrise bei einem Patienten mit zuvor nicht erkanntem nicht-klassischem 21-Hydroxylase-Mangel. In: Intern Med. (2010) 49:1395–400. doi: 10.2169/innere Medizin.49.3573
PubMed Abstract | CrossRef Full Text | Google Scholar
77. Meyer-Bahlburg HFL, Dolezal C, Baker SW, Carlson AD, Obeid JS, New MI. Prenatal androgenization affects gender-related behavior but not gender identity in 5-12-year-old girls with congenital adrenal hyperplasia. Arch Sex Behav. (2004) 33:97–104. doi: 10.1023/B:ASEB.0000014324.25718.51
CrossRef Full Text | Google Scholar
78. Meyer-Bahlburg HFL, Dolezal C, Baker SW, Neu MI. Sexuelle Orientierung bei Frauen mit klassischer oder nicht-klassischer kongenitaler Nebennierenhyperplasie als Funktion des Grades des pränatalen Androgenüberschusses. Arch Sex Spiele. (2008) 37:85–99. doi: 10.1007/s10508-007-9265-1
PubMed Zusammenfassung / CrossRef Volltext / Google Scholar