Blaue Sklera wird durch die Dünnheit und Transparenz der Kollagenfasern der Sklera verursacht, die eine Visualisierung der darunter liegenden Uvea ermöglichen (Abbildung 1). Die Sklera ist der weiße äußere Mantel des Auges, der die Iris umgibt und bei angeborenen Erkrankungen wie Osteogenesis imperfecta ausgedünnt sein kann 1). Andere mit blauer Sklera assoziierte Krankheiten umfassen multiple Bindegewebsstörungen wie das spröde Hornhautsyndrom 2), das Marshall– und Stickler-Syndrom 3), das POEMS-Syndrom (Polyneuropathie, Organomegalie, Endokrinopathie, monoklonales Protein und Hautveränderungen) 4), Marfan-Syndrom, Ehlers-Danlos-Syndrom, Pseudoxanthoma elasticum und Willems-De-Vries-Syndrom, um nur einige zu nennen. Knochen- und Bluterkrankungen, die ebenfalls auf der Liste stehen, umfassen Diamond–Blackfan-Anämie, schwere Eisenmangelanämie, juvenile Paget-Krankheit und sauren Phosphatasemangel 5).Schwere Formen der Osteogenesis imperfecta werden am häufigsten früh im Leben diagnostiziert, aber leichte Fälle können erst später im Leben festgestellt werden. Die blaugraue Farbe der Sklera ist auf die darunter liegenden Aderhautvenen zurückzuführen, die durchscheinen. Dies liegt daran, dass die Sklera dünner als normal ist, da sich das defekte Kollagen Typ I nicht richtig bildet 6). Die Sklera ist eine dichte, schlecht vaskularisierte Bindegewebsstruktur, die aus Kollagen der Typen I, III, IV, V, VI und VIII sowie Elastin, Proteoglykanen und Glykoproteinen besteht 7).In den Vereinigten Staaten wird die Inzidenz von Osteogenesis imperfecta auf eine pro 20.000 Lebendgeburten geschätzt. Schätzungsweise 20.000 bis 50.000 Menschen sind in den USA von Osteogenesis imperfecta betroffen8).Menschen mit Osteogenesis imperfecta werden mit defektem Bindegewebe geboren oder ohne die Fähigkeit, es zu bilden, normalerweise wegen eines Mangels an Kollagen Typ I. Dieser Mangel entsteht durch eine Aminosäuresubstitution von Glycin durch sperrigere Aminosäuren in der Kollagen-Tripelhelix-Struktur. Infolgedessen kann der Körper reagieren, indem er die falsche Kollagenstruktur hydrolysiert 9). Wenn der Körper das unsachgemäße Kollagen nicht zerstört, wird die Beziehung zwischen den Kollagenfibrillen und Hydroxylapatitkristallen zur Bildung von Knochen verändert, was zu Sprödigkeit führt. Als genetische Störung wurde Osteogenesis imperfecta historisch als autosomal dominante Störung des Kollagens Typ I angesehen. Die meisten Fälle wurden durch Mutationen in den Genen COL1A1 und COL1A2 verursacht 10). In den letzten Jahren wurden autosomal-rezessive Formen identifiziert. Mindestens sieben Untergruppen wurden beschrieben, obwohl vier Hauptsubtypen am häufigsten sind und von leicht bis schwer reichen. Personen mit Typ I Osteogenesis imperfecta haben eine geringe Knochendeformität, eine anhaltende blaue Sklera, eine normale Körpergröße im Erwachsenenalter und eine >50% ige Chance auf Hörverlust im Erwachsenenalter. Patienten mit perinataler letaler (Typ II) Osteogenesis imperfecta zeigen die größte Schwere, mit multiplen Frakturen in utero oder von der Lieferung. Diese Patienten sind in der Regel tot geboren oder sterben früh. Die Schwere der Osteogenesis imperfecta hängt vom spezifischen Gendefekt ab. Die meisten Fälle von Osteogenesis imperfecta werden von einem Elternteil vererbt. Einige Fälle sind jedoch das Ergebnis neuer genetischer Mutationen. Eine Person mit Osteogenesis imperfecta hat eine 50% ige Chance, das Gen und die Krankheit an ihre Kinder weiterzugeben 11).
Abbildung 1. Anatomie des Auges
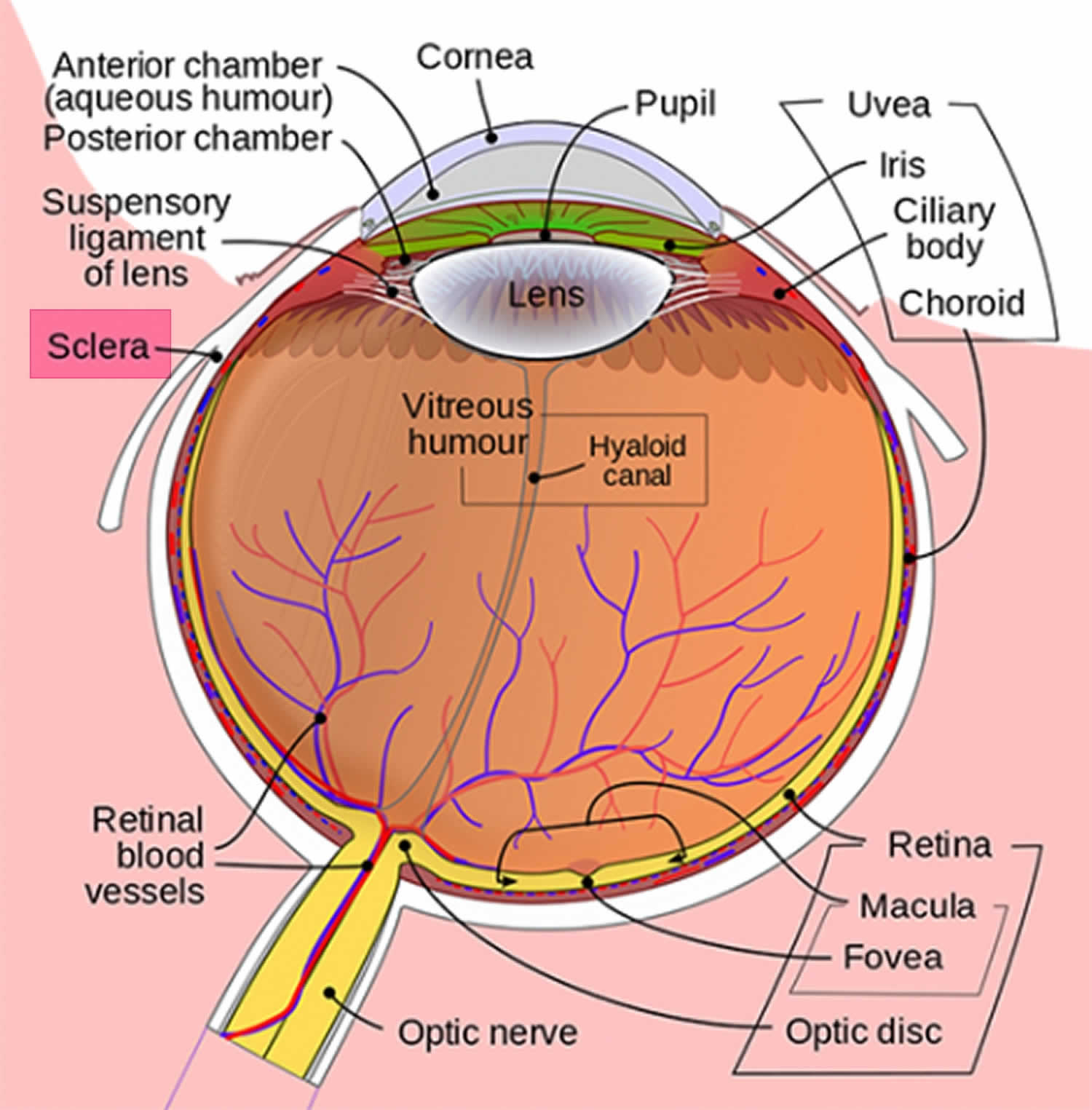
Abbildung 2. Blaue Sklera
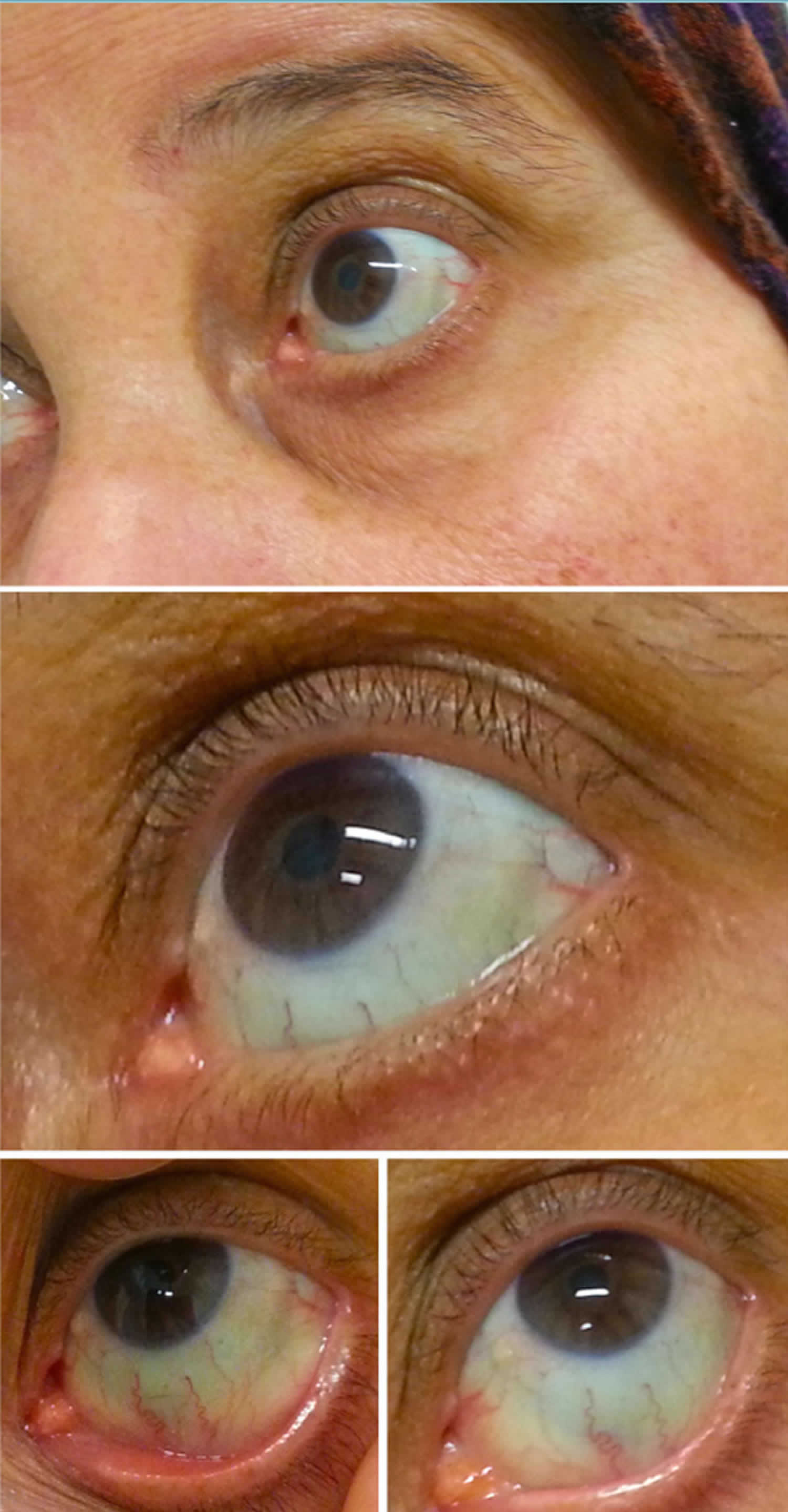
Blaue Sklera verursacht
Blaue Sklera ist die konsistenteste Manifestation der Osteogenesis imperfecta, die aus einer Mutation in COLIA1 und COL1A2 resultiert, die für Typ-I-Prokollagen kodiert. Klassifikationen dieses Zustands (Typen IV–VI) wurden jedoch mit normalen Skleren identifiziert. Osteogenesis imperfecta ist auch mit abnormaler Knochenbrüchigkeit und Taubheit verbunden.Spröde Hornhaut, blaue Sklera und rote Haare sind mit dem spröden Hornhautsyndrom assoziiert, einem Zustand, der auch Skelett-, Zahn- und Hautanomalien aufweist 13). Es wurde festgestellt, dass eine Missense-Mutation in ZNF469 für die Krankheit ursächlich ist.
Andere Grunderkrankungen, die mit der Bildung von blauer Sklera verbunden sind, umfassen das Ehlers-Danlos-Syndrom (Typ VI), Pseudoxanthoma elasticum, Cornea plana, periphere Sklerokornie, Buphthalmus, Keratokonus, Keratoglobus, hohe Myopie, ziliäres / äquatoriales Staphylom, okulodermale Melanozytose und Mikrokornoe. Selten tritt blaue Sklera mit Hallermann-Streiff-Syndrom, Marfan-Syndrom, Turner-Syndrom, Cheney-Syndrom, Menkes-Syndrom, Pyknodysostose, spröder Hornhaut oder ektodermaler Dysplasie auf.Blaue Sklera kann auch bei normalen Säuglingen während der ersten Lebensmonate auftreten; Das Fortbestehen der blauen Verfärbung im Laufe der Zeit kann jedoch auf das Vorhandensein eines erhöhten Augeninnendrucks hindeuten. Frühgeborene zeigen häufig blaue Skleren, insbesondere solche kaukasischer Herkunft.
Blaue Sklera kann auch isoliert als autosomal-dominant vererbte oder autosomal-rezessive Anomalie auftreten 14).
Blaue Sklerasymptome
Blaue Sklera zeigt ein bläuliches Aussehen der Sklera und kann mit pathologischen oder nicht pathologischen Ursachen in Verbindung gebracht werden. Andere Augenmerkmale von Bindegewebsstörungen im Zusammenhang mit blauer Sklera sind dünne Hornhaut, Epikanthal-Falte, Myopie, Keratokonus und Angioid-Streifen. Zu den systemischen Merkmalen von Bindegewebsstörungen im Zusammenhang mit blauer Sklera gehören Hautanomalien, Herzanomalien, Kyphoskoliose, Gelenkhypermobilität, zerbrechliche Knochen, Höranomalien, Gefäßanomalien und gastrointestinale Anomalien 15).
Diagnose der blauen Sklera
Die diagnostische Bewertung der blauen Sklera beinhaltetexterne Untersuchung, Spaltlampenbiomikroskopie und systemische Bewertung für assoziierte Störungen.Obwohl kein definitiver Test für Osteogenesis imperfecta existiert, kann Genetictesting bekannte Mutationen bestätigen oder ausschließen 16).
Behandlung der blauen Sklera
Die Behandlung der blauen Sklera beinhaltet die Diagnose und Behandlung der zugrunde liegenden Ursache. Blaue Sklera ist meist auf genetische Syndrome und in geringerem Maße auf nicht-genetische Störungen zurückzuführen und kann als Nebenwirkung der Medikamenteneinnahme auftreten. Blaue Skleren sind häufig mit angeborenen Störungen der Kollagensynthese verbunden, wie Osteogenesis imperfecta, Marfan–Syndrom, Ehlers-Danlos-Syndrom, Pseudoxanthoma elasticum und Willems-De-Vries-Syndrom, um nur einige zu nennen. Knochen- und Bluterkrankungen, die ebenfalls auf der Liste stehen, umfassen Diamond–Blackfan-Anämie, schwere Eisenmangelanämie, juvenile Paget-Krankheit und sauren Phosphatasemangel 17). Erworbene blaue Skleren wurden bei Erwachsenen mit Augenmelanose, Bindehautmelanom, Alkaptonurie und Addison-Krankheit beschrieben. Geeignete Überweisungen an pädiatrische und orthopädische Untersuchungen können für nicht-okuläre Manifestationen angezeigt sein 18).
Genetische Beratung für assoziierte Erbkrankheiten kann für Patienten mit blauer Sklerose und anderen Manifestationen systemischer Erkrankungen von Vorteil sein.
Bei extremer Ausdünnung und Perforation kann ein chirurgischer Eingriff angezeigt sein. Die strukturelle Unterstützung kann durch ein erhaltenes Skleraltransplantat oder eine autologe Faszie lata bereitgestellt werden, insbesondere in Fällen, in denen ein Nähen einer Vorrichtung wie eines Röhrenimplantats 19 erforderlich ist. Systemische Untersuchungen und Behandlungen sollten in Betracht gezogen werden, um die zugrunde liegende Pathologie anzugehen.
Prognose der blauen Sklera
Die Prognose variiert mit dem Vorhandensein von okulären und systemischen Manifestationen der Grunderkrankung. Patienten mit blauer Sklera haben ein erhöhtes Risiko für Globenruptur oder intraoperative Skleralperforation während routinemäßiger Augenoperationen. Systemische Störungen sind auch bei Patienten wie Karotis-kavernöse Fistel, Arterienruptur, Hörverlust und Knochenbrüche vorherrschend 20).
Referenzen