- definiții și prevalență
- diagnostic
- manifestări NCCAH la femei
- manifestări în copilărie
- manifestări în adolescență și viața adultă
- managementul NCCAH de la prima manifestare la viața adultă
- Managementul în timpul copilăriei
- Management în timpul adolescenței
- pacienți tratați încă din copilărie
- femei cu prima manifestare în timpul adolescenței sau simptome Hiperandrogenice după întreruperea tratamentului
- NCCAH și reproducerea
- subfertilitatea
- avorturi spontane
- planificarea fertilității
- tratamentul Prenatal al CAH
- tratamentul în timpul sarcinii
- alte probleme speciale
- managementul stresului în NCCAH
- aspecte Psihobiologice în NCCAH
- concluzii
- contribuțiile autorului
- Declarație privind conflictul de interese
definiții și prevalență
hiperplazia suprarenală congenitală (CAH) cuprinde o familie de tulburări autosomale recesive caracterizate prin sinteza cortizolului ușor până la acut afectată datorită deficienței uneia dintre cele cinci enzime steroidogene suprarenale necesare pentru producerea cortizolului (1, 2). În mod convențional, CAH este împărțit în (a) clasic (CCAH), prezentându-se cu irosirea sării sau forma virilizantă simplă care se manifestă la naștere și/sau în perioada neonatală și (B) non-clasic (NCCAH), reprezentând o formă mai puțin severă a tulburării care nu are ambiguitate genitală, nu pune imediat viața în pericol și se prezintă mai târziu în viață sau rămâne asimptomatic sau este diagnosticat greșit ca o boală diferită (3). Cu toate acestea, limitele diferitelor forme de CAH nu sunt strict definite, ceea ce tinde să crească provocările asociate cu această tulburare. Prin urmare, este recomandabil să se considere CAH ca un continuum de fenotipuri, de la severă la ușoară sau altfel asimptomatică (3).
cea mai comună formă de CAH este cauzată de deficiența de 21-hidroxilază (21ohd), rezultând o conversie afectată sau deloc de 17 hidroxiprogesteron (17 OHP) la 11 deoxicortizol și de progesteron la deoxicorticosteron. Blocarea conversiei steroizilor are ca rezultat creșterea producției de precursori androgeni, sub stimulare CRH-ACTH, ducând la hiperandrogenism biochimic, marcat de niveluri ridicate de 17-OHP. Forma clasică a bolii, apare la 1 din 16.000 de nașteri vii din întreaga lume. Cu toate acestea, trebuie menționat faptul că incidența percepută a bolii este legată de metoda de screening utilizată. De exemplu, s-a raportat că incidența formei clasice de CAH în Suedia este de 1:11.500 atunci când se utilizează o abordare a studiului de caz, care totuși scade la 1:9.800 atunci când se aplică screeningul hormonal. De asemenea, incidența în Franța și Elveția variază între 1:15.472 și 1:23.000 și 1:10.749 și 1:11.661 atunci când sunt utilizate diferitele metode de screening (4).NCCAH este mult mai frecventă, apărând la aproximativ 1 din 1.000 de caucazieni și mai frecvent la anumite grupuri etnice, cum ar fi evreii Askenazi (1:27), hispanici (1:53), iugoslavi (1:62) și italieni (1:300) (5, 6). Cu toate acestea, lipsesc datele privind prevalența NCCAH pe baza diferitelor metode de estimare (sondaj de caz, screening hormonal și testare moleculară). NCCAH este considerată cea mai frecventă tulburare endocrină autosomală recesivă cu o frecvență purtătoare de 1:25 până la 1:10. În NCCAH datorită 21-OHD, activitatea enzimatică reziduală este estimată la aproximativ 10-70% (7-9).
diagnostic
în comparație cu diagnosticul formei clasice a bolii, care se face la naștere sau în perioada neonatală din cauza ambiguității genitale și / sau a simptomelor de risipă de sare sau prin programe de screening utilizate în unele țări, majoritatea cazurilor de NCCAH nu sunt ușor detectabile (4, 10). În plus, mulți indivizi rămân asimptomatici în timpul copilăriei și adolescenței, au o funcție de reproducere normală și devin conștienți de NCCAH numai datorită diagnosticului unui alt membru al familiei și testării ulterioare (11). Cu toate acestea, majoritatea femeilor cu NCCAH solicită asistență medicală atunci când prezintă simptome de exces de androgeni și, atunci când suspiciunea clinică solicită testarea, nivelurile crescute de 17 OHP bazale vor indica mai mult decât un diagnostic de NCCAH.
într-adevăr, ghidurile clinice propuse de Societatea Endocrină recomandă o valoare de bază non-stimulată de 17 OHP ca instrument de screening pentru NCCAH. Dimineața 17 niveluri OHP>6 nmol/l în faza foliculară la femeile menstruale ar trebui să solicite o evaluare suplimentară, deoarece s-a demonstrat că nivelurile peste 6 nmol/l captează 90% din indivizii NCCAH (12, 13). Măsurătorile aleatorii de 17 OHP nu s-au dovedit a fi utile, deoarece acestea produc adesea niveluri normale la pacienții cu NCCAH și sunt, în plus, extrem de ridicate în faza luteală la jumătate din femelele sănătoase (13). Cu toate acestea, în datele noastre derivate din 280 de subiecți cu boala, șase pacienți (2.1%) a avut o valoare inițială de 17 OHP < 6 nmol/L (14). În plus, Bidet și colab., într-o cohortă mare de femei cu NCCAH verificate prin tehnici moleculare, s-au găsit, de asemenea, valori bazale de 17 OHP mai mici de 6 nmol/L la 8% dintre subiecții studiați (15). În cele din urmă, pe baza datelor colectate de Speiser și colab., 9% dintre persoanele cu NCCAH au prezentat 17 valori OHP mai mici de 2 ng/ml (ceea ce corespunde la 6nmol/L). Conform altor studii, o valoare de bază de 17 OHP între 5,1 și 9 nmol/L este suficientă pentru diagnosticul NCCAH (13, 16, 17). Recent, un nivel de bazal 17 OHP de 4.6 nmol / l a fost sugerat ca un prag pentru testarea ACTH pentru a prezice NCCAH la subiecții cu adrenarche prematură în timpul copilăriei (18).
suma totală a acestor constatări și sugestii indică faptul că selecția pacienților care vor fi supuși unui test de stimulare Synacthen trebuie evaluată de la caz la caz. Pentru diagnostic este necesar un nivel post-stimulare de 17 OHP de peste 3 nmol / L (19). Heterozigoții care poartă o mutație CYP21A2 prezintă niveluri ușor crescute de 17 OHP după stimularea ACTH, deși există suprapuneri la subiecții neafectați (9). Cu toate acestea, ca Dacou-Voutetakis și colab. au subliniat, dacă suma valorilor bazale și post-stimulare 17 OHP depășește 1,5 nmol / L, atunci posibilitatea heterozigozității este excepțional de mare (20).
un alt aspect important se referă la tehnicile utilizate pentru evaluarea 17 OHP. 17 niveluri OHP sunt măsurate printr-o varietate de metode de imunotestare, dar după cum sa arătat recent, cele mai precise și fiabile rezultate au fost obținute prin implementarea combinației de cromatografie lichidă cu spectrometrie de masă (LC-MS/MS). Într-adevăr, multe măsurători fals pozitive 17 OHP au fost găsite atunci când măsurătorile LC-MS/Ms au fost comparate cu metodele standard (21) Cu toate acestea, ultimele proceduri nu sunt utilizate universal, în cazul unui diagnostic incert, acestea vor oferi rezultate mai precise. De remarcat, pragurile de screening și diagnostic pentru LC-MS/MS cuantificate 17 OHP nu sunt încă definite. Aceste praguri sunt probabil mai mici decât cele stabilite cu imunoteste datorită specificității sporite a LC-MS/MS, o metodă mai puțin predispusă la reactivitate încrucișată și interferențe (22, 23). Rămâne de elucidat dacă este necesar un profil steroid urinar pentru diagnosticul definitiv (22). În cazurile limită, se recomandă obținerea unui profil adrenocortical complet după testul de stimulare ACTH pentru a diferenția deficiența de 21-hidroxilază de alte defecte enzimatice și pentru a stabili un diagnostic ferm.
în mod specific, trebuie efectuat un profil complet de steroizi în cazuri echivoce pentru a confirma deficiența de 21-hidroxilază și pentru a exclude alte detectări enzimatice. Includerea a 17 OHP, cortizol,11-deoxicorticosteron,11-deoxicortizol, 17-OH-pregnenolonă, dehidroepiandrosteron și androstendionă într-un profil seric de steroizi ar fi utilă pentru a exclude alte cauze ale Cah, cum ar fi deficitul de 11-hidroxilază și, mai rar, deficitul de 3-hidroxisteroid dehidrogenază sau deficitul de oxidoreductază P450 (24). În timp ce testarea genetică nu este considerată a fi un instrument de diagnostic primar pentru NCCAH în acest moment, este obligatorie pentru confirmarea diagnosticului și pentru consilierea genetică (3).
în ceea ce privește nivelurile de cortizol, în general se așteaptă o valoare post-stimulare de cel puțin 496 nmol/l. De remarcat, potrivit Stoupa și colab., 60% din 47 de copii cu NCCAH ca urmare a 21 OHD au avut valori scăzute ale cortizolului după stimulare, o constatare care indică necesitatea unei supravegheri sporite pentru dezvoltarea insuficienței suprarenale în timpul evenimentelor majore de stres (25).
manifestări NCCAH la femei
expresia clinică a NCCAH se caracterizează printr-un nivel ridicat de polimorfism în ceea ce privește nu numai vârsta de debut, ci și diferitele semne și simptome. Este raportat că prima prezentare clinică a NCCAH este în 11% din cazuri înainte de vârsta de 10 ani și în 80% între vârstele de 10 și 40 de ani (12). Corelația genotip-fenotip în CAH și NCCAH nu a fost încă elucidată. Speiser și colab. sugerează că majoritatea, dar nu toate, variabilitatea fenotipică a deficitului de 21-hidroxilază rezultă din variația alelică a CYP21A2 (26).
manifestări în copilărie
în perioada nou-născutului, de obicei femelele NCCAH rămân asimptomatice și au organe genitale externe normale. Cel mai vechi caz de NCCAH raportat este o fetiță de 6 luni care a dezvoltat părul pubian (11). De obicei, constatările clinice și simptomele în cazurile NCCAH încep de la vârsta de 5 ani sau chiar mai târziu și sunt legate de creșterea nivelului de androgeni, deși deficitul ușor de cortizol poate apărea, de asemenea, în unele cazuri (8). În 92% din cazurile NCCAH, primul simptom care se manifestă este pubarche prematură (PP), care apare sub 8 ani la fete și sub 9 ani la băieți (12) cu o prevalență estimată între 10 și 11,3% (15). De remarcat, la copiii menționați cu PP, conform diferitelor studii, incidența NCCAH variază de la 5 la 30% (20, 27). Cu toate acestea, pe baza analizei noastre a 280 de pacienți cu confirmare moleculară a NCCAH, am constatat că incidența PP la 94 de femei cu vârsta sub 8 ani a fost de până la 88%. Pe de altă parte, o analiză a 45 de bărbați cu NCCAH a identificat PP la doar 29% dintre subiecți (14). Mai mult, ar trebui să ținem cont de faptul că aproximativ jumătate dintre subiecții cu PP pot fi purtători heterozigoți ai unei mutații CYP21A2.
Un alt aspect care necesită clarificări este relația dintre PP și pubertatea precoce. Se presupune că, în unele cazuri, aromatizarea periferică a androgenilor suprarenali la estrogeni poate activa axa hipotalamo-hipofizo-gonadală, ducând la pubertate precoce secundară (28). Într-un astfel de caz, este necesară o monitorizare regulată. Unii copii dezvoltă hirsutism, acnee și/sau creștere rapidă și manifestă o structură înaltă și o avansare a vârstei osoase (29). Aceasta din urmă poate duce la înălțimea finală trunchiată ca o consecință a fuziunii epifize rapide. Alte descoperiri clinice rare în timpul copilăriei includ adeziunea labială, părul perianal, clitoromegalia și răgușeala vocii (30-32).
manifestări în adolescență și viața adultă
în timpul adolescenței și maturității, NCCAH la femei prezintă simptome hiperandrogenice asemănătoare sindromului ovarului polichistic (PCOS) (33). Simptomele includ hirsutism, acnee, alopecie androgenică, anovulație, disfuncție menstruală și infertilitate. Într-un studiu multicentric, cele mai frecvente simptome în rândul femeilor adolescente și adulte au fost hirsutismul (59%), oligomenoreea (54%) și acneea (33%). Dintre cele 161 de femei cu NCCAH, care au prezentat simptome au fost hirsutismul (78%), disfuncția menstruală (54,7%) și scăderea fertilității (12%) (34). În grupul nostru de 161 de femei cu boala, 63% dintre pacient au prezentat un fenotip asemănător ovarului polichistic (14). Această constatare are o implicație invers, deoarece incidența NCCAH este ridicată la femeile cu hiperandrogenemie. Desigur, studiul lui Witcel și colab. raportarea unei incidențe a NCCAH cuprinsă între 1 și 33% la femeile hiperandrogenice este de mare interes. Într-un alt studiu efectuat pe 950 de femei care au fost trimise pentru evaluarea excesului de androgeni, NCCAH a fost detectat la 4,2% dintre ele. Cu toate acestea, pacienții NCCAH au fost mai tineri și mai hirsuți în comparație cu celelalte grupuri de pacienți hiperandrogenici. Acestea au fost, de asemenea, caracterizate de niveluri semnificativ mai ridicate de testosteron, testosteron liber și 17 OHP decât pacienții cu alte sindroame hiperandrogenice. În ecografia ovariană, 77% dintre aceștia au prezentat ovare polichistice și 41% au crescut dimensiunea ovariană. În concluzie, acestea au îndeplinit criteriile PCOS în conformitate cu NIH și Rotterdam la un procent de 56% și, respectiv, 72,8% (35).
natura progresivă a bolii este evidențiată de faptul că prevalența hirsutismului sa dovedit a crește odată cu vârsta și a fost observată a fi rară înainte de pubertate. Hirsutismul este cea mai frecventă manifestare clinică raportată la pacienții cu NCCAH, variind de la 71 la 96% (28, 36). Fetele pubertale cu NCCAH prezintă de obicei hirsutism (37, 38). La celălalt capăt al spectrului este problema alopeciei. La pacienții cu NCCAH s-a raportat chelire de tip masculin. Prevalența alopeciei a apărut, de asemenea, să crească odată cu vârsta, de la 6% la pacienți în a doua decadă a vieții lor la 19% în a cincea, indicând din nou natura progresivă a bolii (39). Acneea este raportată la aproape 33% dintre subiecții NCCAH (36). În mod remarcabil, mutațiile genei CYP21A2 care cauzează NCCAH au fost detectate la 4,9% din 123 de femei adulte care prezintă acnee severă (40).
neregularitățile menstruale, inclusiv oligomenoreea sau amenoreea secundară, pot fi adesea semnul prezentator al NCCAH la persoanele post-menarche. În plus, 8-9% din cazuri prezintă, de asemenea, amenoree primară și, din acest motiv, solicitați sfatul medicului pentru prima dată. Mai mult, printre indivizii NCCAH, oligomenoreea este mai frecventă decât amenoreea primară. De exemplu, într-o serie de la Moran și colab., printre femeile NCCAH cu vârsta cuprinsă între 10 și 19 ani, oligomenoreea a fost prezentă în 56% din cazuri, comparativ cu doar 9% din adolescență cu amenoree primară (12). În concluzie, atât de multe simptome NCCAH seamănă cu cele ale PCOS, încât nu este surprinzător faptul că NCCAH a fost supranumit Marele mimicker al PCOS. În orice caz, un diagnostic de NCCAH trebuie luat în considerare în timpul evaluării oricărei tinere care este trimisă pentru simptome hiperandrogenice.
într-un studiu realizat de Arlt și colab., pacienții cu NCCAH au fost caracterizați prin IMC semnificativ mai mare și înălțime finală mai mică în comparație cu controalele potrivite (41). Aceste date, coroborate cu câteva studii care indică prezența insensibilității la insulină, sugerează un profil metabolic nefavorabil care ar putea fi explicat fiziopatologic prin efectul concentrațiilor crescute de androgeni și/sau ca urmare a terapiei cu glucocorticoizi (42, 43). Osteoporoza poate fi, de asemenea, detectată la acești subiecți, probabil ca o consecință a terapiei cu corticosteroizi (34).
managementul NCCAH de la prima manifestare la viața adultă
odată ce diagnosticul NCCAH a fost stabilit, mai multe aspecte legate de recomandările ulterioare de tratament justifică luarea în considerare. Prima întrebare care trebuie abordată este dacă este indicată terapia cu glucocorticoizi (GC). Raționamentul general din spatele acestei abordări este că, oferind pacientului concentrații suficiente de cortizol, nevoile sale zilnice sunt acoperite și, în consecință, stimularea axei CRH-ACTH va fi conică, ducând la scăderea producției de androgeni suprarenali. Regula generală referitoare la această abordare este că GCs se administrează la doze de înlocuire și nu farmacologice, în timp ce influența vârstei, sexului, datelor de laborator, recomandărilor specifice pacientului și obiectivelor tratamentului asupra terapiei de substituție cu glucocorticoizi sunt luate în considerare în mod serios. Cu toate acestea, ar trebui să fim conștienți de faptul că terapia de substituție GC prelungită poate duce la hipercortizolemie, cu efecte adverse bine documentate asupra fiecărui aspect al metabolismului, în special creșterea, distribuția grăsimilor și rezistența la insulină, precum și asupra profilului psihologic. Un alt dezavantaj major al acestei abordări este lipsa unui indice clinic adecvat sau a unui marker biochimic al dozei adecvate de înlocuire, cum ar fi valorile TSH în hipotiroidism.
în cele din urmă, faptul că nu există un consens cu privire la care GC ar trebui utilizat și absența datelor pe termen lung cu privire la diferitele modalități de administrare a GC complică și mai mult deciziile medicilor participanți și selectarea alegerii optime pentru fiecare pacient. Hidrocortizonul este utilizat de obicei la copii, deoarece seamănă cel mai mult cu hormonul natural (cortizolul), dar nu este considerat o abordare adecvată la adolescenți și femei tinere din cauza necesității dozării zilnice multiple. Prin urmare, majoritatea endocrinologilor adulți preferă fie dexametazona, fie prednisolonul, la doze adecvate. Pe de altă parte, trebuie subliniat faptul că echivalența diferitelor GCs se bazează pe acțiunea lor antiinflamatorie și nu pe diferite aspecte ale metabolismului uman. Deoarece cortizolul afectează aproape 20% din genomul uman, sunt așteptate răspunsuri diverse ale diferitelor GCs în diferite țesuturi. Coroborarea acestei percepții este faptul că administrarea dexametazonei la pacienții cu CAH a arătat deteriorarea indicilor de rezistență la insulină în comparație cu alte GCs (44). În plus, administrarea a 2,5-7,5 mg prednisolon, o doză considerată normală, exercită un impact negativ de lungă durată asupra metabolismului osos (45). Prin urmare, hidrocortizonul trebuie considerat cea mai bună formă de tratament în cazurile de terapie suplimentară cu GC. Apariția formulei de hidrocortizon nou sintetizate cu o pastilă pe zi și rezultatele sale pozitive inițiale la pacienții cu CAH arată multă promisiune pentru viitor (46).
Managementul în timpul copilăriei
pacienții NCCAH care sunt diagnosticați în timpul copilăriei cu semne de PP pot fi tratați cu hidrocortizon cu scopul de a suprima hormonii suprarenali și de a preveni avansarea rapidă a vârstei osoase care ar putea afecta înălțimea finală. La acei pacienți la care tratamentul cu hidrocortizon a fost inițiat cu 1 an înainte de debutul pubertății și care aveau o vârstă osoasă sub 9 ani, înălțimea finală a rămas în limitele potențialului genetic (47). Studiile disponibile indică faptul că înălțimea adulților s-a apropiat de înălțimea țintă așteptată la pacienții care au fost monitorizați îndeaproape și care au respectat cu strictețe planurile de medicație (48-50). Un mic studiu recent efectuat pe cinci cazuri (pacienți cu vârsta cuprinsă între 6,1 și 9,2 ani) a demonstrat că o doză ultralow de dexametazonă este o opțiune promițătoare pentru reducerea stresului endogen și a efectelor acestuia. Rămâne de elucidat dacă aceasta constituie o abordare realistă și cât de mult trebuie administrată doza (51). Un studiu realizat de Nebesio și colab. a comparat efectele hormonale și farmacocinetica hidrocortizonului, prednisolonului și dexametazonei la 9 pacienți prepubertali cu CCAH. Ei au arătat că dexametazona a fost mai puternică decât celelalte forme în atingerea unor niveluri semnificativ mai scăzute ale hormonilor suprarenali, sugerând astfel că dexametazona este mai eficientă pentru suprimarea producției de androgeni suprarenali (46). Desigur, sunt necesare studii suplimentare pentru a verifica sau respinge această constatare. În plus, în unele cazuri, concentrațiile crescute de androgeni pot duce la stimularea secundară a axei GnRH, ducând la pubertate prematură. Într-un astfel de caz, poate fi prescris un curs paralel cu analog GnRH dacă vârsta osoasă este semnificativ mai mare decât vârsta cronologică și/sau înălțimea finală proiectată este disproporționată față de înălțimea țintă.
Un alt aspect important se referă la cazurile de deficiență concomitentă de GH în cazul în care tratamentul cu analogi GnRH și prescrierea GH a dus la atingerea înălțimii țintă (52). Conform ghidurilor actuale, terapia descrisă mai sus este recomandată cu precauție numai pentru acele cazuri în care înălțimea prezisă SD este -2,25 < înălțimea țintă sau chiar mai mică (53). O indicație alternativă pentru începerea tratamentului cu hidrocortizon este un răspuns inadecvat al cortizolului după stimularea ACTH (36).
tratamentul GC preferat la copii este de obicei hidrocortizonul 10-15 mg / m2, împărțit în trei doze. Adesea, dozele mai mici s-au dovedit eficiente, începând de la 6 mg/m2/zi (34). Supradozajul trebuie evitat, având în vedere că poate duce la o creștere slabă, precum și la caracteristici Cushingoide. Modelul de creștere regulat, o vârstă osoasă compatibilă cu vârsta cronologică și absența obezității centrale pot servi, de asemenea, ca indici clinici pentru un management adecvat. În ceea ce privește profilul biochimic/ hormonal, nivelurile de androstendionă și testosteron în intervalele medii până la superioare pentru vârsta osoasă sunt considerate markeri mai buni ai terapiei adecvate de substituție GC la copiii cu NCCAH. Cu toate acestea, nivelurile de testosteron suprimate au fost găsite la 10% dintre pacienții cu NCCAH, în timp ce alți 28% dintre pacienți au avut concentrații crescute de testosteron, acest fenomen putând fi atribuit variabilității hidrocortizonului (54, 55). Valorile DHEAS sunt scăzute cu doze foarte mici, în timp ce suprimarea nivelurilor de 17 OHP și progesteron necesită doze foarte mari de GC. Trebuie remarcat faptul că valorile 17 OHP sunt cruciale pentru diagnostic, dar nu sunt utile în timpul urmăririi. De remarcat, prelevarea de sânge pentru evaluarea hormonală trebuie efectuată fără întreruperea tratamentului. Cu toate acestea, din cauza nedumeririi bolii și a naturii sale multiple, nu există linii directoare specifice pentru momentul modificărilor regimului sau încetarea terapiei cu glucocorticoizi la copii.
Management în timpul adolescenței
pacienți tratați încă din copilărie
până la stabilirea modelului menstrual normal la fetele NCCAH, continuarea GCs care a început în timpul copilăriei este foarte recomandată (56). Cu toate acestea, pacienții adolescenți nu prezintă frecvent o respectare suficientă a administrării cronice a medicamentelor și adesea omit dozele. În plus, în timpul pubertății, timpul de înjumătățire al hidrocortizonului scade cu 50% ca urmare a nivelurilor crescute de IGF-1, care diminuează activitatea de 11 centicohsd, precum și datorită creșterii clearance-ului cortizolului care rezultă din amplificarea ratei de filtrare glomerulară (57). La femela tânără, la 2 ani după menarche și dacă au fost înregistrate cicluri ovulatorii normale, se recomandă o abordare centrată pe pacient față de simptomele hiperandrogenice care pot apărea.
dacă există constatări hiperandrogenice severe, cum ar fi hirsutismul, acneea și / sau oligomenoreea, se va lua în considerare continuarea tratamentului. Este important să ne amintim sensibilitățile fragile ale unui adolescent tânăr, care vor fi grav afectate de semnele „respingătoare” ale hiperandrogenismului. Combinația dintre semnele hiperandrogenice, tulburările menstruale și calitatea slabă a vieții este bine documentată și este deosebit de ridicată la femeile mai tinere. Un alt punct care trebuie luat în considerare este răspunsul stimulării post ACTH a rezervei suprarenale. Având în vedere că cortizolul maxim al femelelor pubertale și adulte după stimulare este sub 496 nmol/L, în cazurile de stres, trebuie administrat tratament cu steroizi (58). Pe de altă parte, dacă niciunul dintre simptomele de mai sus nu este întâlnit la pacientul tânăr, tratamentul cu GC poate fi întrerupt, după care se recomandă urmărirea regulată.
femei cu prima manifestare în timpul adolescenței sau simptome Hiperandrogenice după întreruperea tratamentului
în prezența simptomelor hiperandrogenice, este foarte recomandată o abordare orientată spre pacient, concentrându-se pe principalele plângeri ale pacientului. Acest lucru este mult mai bun decât un modus operandi universal general, deoarece, cu această din urmă abordare, tânărul pacient va fi dezamăgit, ceea ce va duce la întreruperea tratamentului și, ca urmare, la rezultate negative asupra sănătății la vârsta adultă. În cazurile cu hirsutism sever, un curs de 6 până la 12 luni cu pilule contraceptive orale (OCP) constituie o procedură rezonabilă, având în vedere efectele benefice ale OCP asupra producției hepatice SHBG și scăderea eliberării de androgeni din ovar. Îmbunătățirea clinică va fi așteptată după cel puțin 2-3 luni de la inițierea OCP, în timp ce utilizarea concomitentă a unui progestagen cu proprietăți androgenice minime este foarte recomandată. Dacă OCP eșuează ca abordare de primă linie, antiandrogenii (spironolactonă, flutamidă, bicalutamidă, acetat de ciproteronă și finasteridă) pot fi adăugați la tratament. Din experiența noastră, administrarea de bicalutamidă a obținut o îmbunătățire semnificativă în cazurile de acnee severă, dar rezultate similare nu au fost obținute în cazurile cu hirsutism sever. Abordările cosmetice, cum ar fi aplicarea cu laser și depilatoarele, pot fi, de asemenea, sugerate femeilor care se plâng de creșterea excesivă sau nedorită a părului sau de căderea părului scalpului (alopecie androgenică) (32, 34, 57).
la pacienții cu secreție inadecvată de cortizol după stimulare sau dacă OCP și antiandrogeni nu pot fi tolerați, se recomandă un curs cu GCs (57). Date de la New și colab. indicați că menstruația neregulată și acneea sunt inversate în decurs de 3 luni de la inițierea terapiei cu glucocorticoizi, în timp ce hirsutismul necesită aproape 30 de luni (59). Spre deosebire de copilărie, în adolescență se utilizează adesea steroizi cu acțiune mai lungă și se recomandă regimuri de 5 mg prednisolon sau 0,25 mg dexametazonă (53). Cu toate acestea, în lumea reală datele clinice au arătat o varietate de regimuri diferite aplicate în managementul NCCAH (41).
scopul principal al tratamentului ar trebui să fie scutirea pacientului de simptomele hiperandrogenice. Astfel, în rândul persoanelor cu NCCAH diagnosticate exclusiv pe baza rezultatelor de laborator, caracteristicile clinice ar trebui să ghideze recomandările de management, deoarece constatările hormonale și moleculare nu prezic neapărat severitatea clinică. Conform ghidurilor Societății Endocrine, pacienților cu NCCAH trebuie să li se ofere opțiunea de a întrerupe tratamentul cu GC atunci când simptomele dispar (60). Desigur, acești pacienți nu trebuie pierduți la urmărire, în timp ce tratamentul trebuie reluat în cazul reapariției simptomelor. În plus, în cazul întreruperii tratamentului, pacienții trebuie informați cu privire la posibilitatea infertilității și trebuie încurajați să solicite sfatul medicului dacă doresc să conceapă (19). De remarcat, transferul adecvat al pacientului de la pediatru la endocrinologul adult trebuie efectuat, în mod optim după 1 an de monitorizare sincronizată (53, 61).
NCCAH și reproducerea
subfertilitatea
subfertilitatea este frecvent raportată în formele clasice de CAH, care se datorează în principal tulburărilor menstruale, anovulației cronice și deformărilor anatomice. Rata natalității a fost estimată la 17% în comparație cu 65% din populația de control (62). Între timp, datele privind fertilitatea la femeile cu NCCAH au fost evaluate recent în detaliu, iar incidența estimată a infertilității este de 11% în rândul femeilor NCCAH, adică relativ mai ușoară decât în CAH și, în multe cazuri, este ușor de rezolvat. Bidet și colab. a evaluat fertilitatea la 190 de femei care suferă de NCCAH, dintre care 95 doreau să rămână însărcinate. În această populație, au fost raportate 187 DE SARCINI la 85 de femei, ceea ce a dus la 141 de nașteri la 82 de persoane. Trebuie subliniat faptul că 99 dintre SARCINI (52,9%) au avut loc înainte de diagnosticul NCCAH, trei dintre ele cu aplicarea protocoalelor de inducție a ovulației, restul fiind spontane, în timp ce 88 de sarcini au avut loc după diagnosticul NCCAH. În marea majoritate a acestora, sarcina sa dezvoltat după instituirea terapiei cu hidrocortizon, în timp ce la 11 femei sa întâmplat spontan (63, 64). Într-un alt raport al 22 de femei NCCAH observate care doresc sarcină, 12 sarcini au urmat cu prednison (65).
avorturi spontane
în cohorta de Bidet și colab., rata de avort spontan a fost de 6, 5 și 26, 3% la pacienții tratați cu GCs și, respectiv, la pacienții netratați (64). Rezultatul sarcinii poate avea chiar succes fără niciun tratament cu glucocorticoizi în cazurile în care NCCAH nu a fost încă diagnosticat, așa cum s-a raportat și într-un raport de caz realizat de Cuhaci și colab. Cu toate acestea, același cuplu a suferit două avorturi spontane și a raportat subfertilitate după această primă sarcină (66). În ceea ce privește posibilitatea sarcinilor premature, evaluată de Bidet și colab., nu diferă semnificativ de cea a restului populației (15).
planificarea fertilității
pentru femeile cu hiperandrogenism simptomatic sau cu infertilitate raportată, dar care doresc să conceapă, terapia GC este foarte recomandată. În această perioadă, glucocorticoizii utilizați sunt prednison sau hidrocortizon. Cea mai importantă acțiune, deoarece va ghida pașii următori, este testarea genetică a viitorilor părinți. Femeile cu NCCAH care doresc să conceapă ar trebui să fie conștiente de riscul de a naște un copil cu forma clasică a bolii. Rezultatul sarcinilor în rândul femeilor cu NCCAH și, mai precis, incidența sugarilor născuți cu CCAH, este estimat în studii recente între 1,5 și 2,5%, cu NCCAH la aproximativ 15% (36, 64). Desigur, această frecvență depinde de populația de referință, o incidență mai mare apărând la populațiile cu rate mari de căsătorie (36). Șansele prezise ca părinții să nască un copil cu CAH sunt 1 din 120 pentru genotipul patern necunoscut, zero dacă tatăl nu are mutație relevantă și 1 din 4 dacă are o mutație heterozigotă (19). Ca urmare, testarea genetică a partenerilor acestor femei este esențială pentru a evalua riscul nașterii unui copil cu forma clasică de CAH (64, 67).
în cazul în care mama potențială poartă două mutații NC (Tabelul 1), nu este nevoie absolută de testare genetică la viitorul tată, deoarece, dacă este heterozigot pentru mutațiile CYP12A2, descendenții sunt expuși riscului de a dezvolta forma non-clasică a bolii în viitor. Cu toate acestea, trebuie remarcat faptul că statutul de purtător este de 2-15% în diferite populații și jumătate dintre acești indivizi poartă o mutație severă. Mai mult, recomandările definitive privind situațiile în care testarea genetică nu este necesară sunt dificile, având în vedere corelația imperfectă genotip-fenotip, în special pentru mutații mai ușoare . De exemplu, mutația P30L, cel mai frecvent asociată cu NCCAH, apare și la pacienții cu CAH clasic . Un studiu european multicentric mare a arătat recent că acesta a fost cazul la 78% dintre pacienți . În acest studiu, mutația ușoară V218L a fost asociată cu Clasic mai degrabă decât cu NCCAH în 30% din cazuri. Prin urmare, tuturor pacienților cu NCCAH ar trebui să li se ofere consiliere genetică și evaluare moleculară a partenerilor de reproducere.
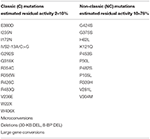
Tabelul 1. Mutații ale CYP21A2 cu activitate enzimatică reziduală minimă (C) sau moderată (NC) (14, 68, 69).
în schimb, în cazul în care mama potențială este un heterozigot compus cu o mutație severă (C) și o mutație NC, atunci testarea genetică a viitorului tată este obligatorie. Ar trebui să ținem cont de faptul că datele provenite din studii care utilizează imunoteste sau procedurile LC-MS/MS mai precise, evidențiază în mod constant incapacitatea de a exclude în mod fiabil heterozigozitatea folosind valori bazale sau ACTH stimulate 17 OHP, datorită suprapunerii semnificative cu intervalul normal. Se poate sugera utilizarea unui test Synacthen și, dacă nu este compatibil cu heterozigozitatea (suma valorilor bazale și maxime stimulate 17 OHP < 1,5 nmol/L), atunci testarea ADN ar putea fi evitată. Cu toate acestea, ar trebui să fim foarte precauți în interpretarea acestui test, deoarece nu a fost validat în alte studii. Prin urmare, utilizarea ACTH stimulat 21-deoxicortisol fie singular sau ca parte a unui raport de steroizi sau profil de steroizi, poate facilita identificarea biochimică a heterozigoților în viitor, în special pe măsură ce LC-MS/MS devine mai disponibil pe scară largă . Dacă tatăl potențial poartă o mutație NC, atunci nu este nevoie de nimic altceva. În schimb, dacă este un purtător al unei mutații clasice, va apărea întrebarea privind tratamentul prenatal al fătului. Toate combinațiile posibile de genotipuri și procedurile sugerate sunt prezentate în tabelul 2.

Tabelul 2. Planificarea procedurilor sugerate în timpul sarcinii pe baza genotipului părinților potențiali.
tratamentul Prenatal al CAH
scopul tratamentului prenatal al CAH este prevenirea virilizării genitale a fătului, dar și atenuarea stresului resimțit de părinții care sunt susceptibili de a avea un copil cu organe genitale ambigue (70). Dexametazona este utilizată datorită capacității sale de a traversa placenta și pentru că nu este dezactivată de la steroidul dehidrogenază placentară 11 Oh și se leagă doar minim de globulina care leagă cortizolul din sângele mamei (CBG). Dexametazona, prin suprimarea secreției fetale de ACTH, scade producția crescută de androgeni fetali. Tratamentul trebuie întrerupt în cazul în care cariotipul sau analiza ADN relevă un bărbat sau, respectiv, o femeie neafectată (71). Cu toate acestea, deși virilizarea genitală fetală a început deja la 6-7 săptămâni după concepție, biopsiile viloase corionice pentru diagnosticul genetic nu pot fi obținute înainte de săptămâna 10-12. Acest interval de timp sugerează că toate sarcinile cu risc de virilizare a CAH trebuie tratate, chiar dacă doar 1 din 8 fetuși este afectat și feminin (19, 34, 70).
dacă această expunere a fătului la dexametazonă pentru măsuri preventive este acceptabilă din punct de vedere medical și etic rămâne controversată (70). Studiile la animale au demonstrat că dexametazona din primul trimestru scade greutatea la naștere, afectează funcția celulelor beta pancreatice renale și dezvoltarea creierului și predispune la anxietate, hipertensiune arterială și hiperglicemie la vârsta adultă (70). Mai mult, 10-20% dintre femeile însărcinate care utilizează tratamentul cu dexametazonă în timpul sarcinii se plâng de creșterea în greutate, creșterea poftei de mâncare, modificări ale dispoziției, insomnie, edeme, hipertensiune arterială, hiperglicemie și striae. Cu toate acestea, New și colab. s-a constatat un scor Prader mai mic decât media la fetușii tratați cu dexametazonă prenatal, dar nici o diferență în rezultatele pe termen lung (72). În plus, într-o recenzie de Merce Fernandez-Balsells și colab., dexametazona s-a dovedit a fi asociată cu reducerea virilizării fătului fără efecte adverse materne sau fetale semnificative (63). Cu toate acestea, autorii revizuirii menționate mai sus, precum și orientările consecutive ale Societății Endocrine subliniază că, din cauza dimensiunilor mici ale eșantionului din întregul corp al literaturii, subiectul rămâne incert și este în mod clar necesară o investigație suplimentară.
decizia privind inițierea tratamentului trebuie luată numai în centre mari, cu o echipă experimentată și protocoale aprobate de comisiile de evaluare instituționale și bazate pe valorile și preferințele familiei și cu acordul lor scris informat ca o condiție prealabilă (19, 63). Mai mulți specialiști în domeniu indică faptul că ar trebui acordată o valoare mai mare prevenirii expunerii prenatale inutile a mamei și fătului la dexametazonă, mai degrabă decât impunerea taxei emoționale a organelor genitale ambigue asupra părinților și pacienților. Cel mai important, trebuie clarificat părinților că administrarea dexametazonei nu Modifică starea pacientului, ci este îndreptată spre reducerea nevoii de intervenție chirurgicală, mai degrabă decât spre păstrarea vieții sau a capacității intelectuale. De asemenea, trebuie să știe că probabilitatea ca copilul lor să sufere de forma clasică a bolii este mare, în ciuda tratamentului. Cel mai important, între timp, expunerea unui organism tânăr la un GC foarte puternic în timpul unei perioade deosebit de sensibile de programare și creștere fetală, care s-ar putea dovedi inutilă în cazul unui făt cu cariotipul XY, nu este în prezent susținută de date robuste și incontestabile.
în mod ideal, ar trebui să existe un diagnostic precoce, acest lucru efectuat printr-o tehnică neinvazivă și înainte de începerea virilizării genitale a fătului. Lucrând în această direcție, studiile noi indică utilizarea ADN-ului fetal fără celule obținut din plasma maternă ca metodă promițătoare care va permite determinarea sexului fetal și diagnosticarea CAH la o vârstă gestațională timpurie (<9 săptămâni). Dacă această procedură este implementată pe scară largă în practica clinică, tratamentul prenatal inutil cu dexametazonă va fi evitat (73).
tratamentul în timpul sarcinii
în timpul sarcinii, femeile cu NCCAH ar trebui, de preferință, tratate cu hidrocortizon, nu numai pentru că sarcina este însoțită de obicei de niveluri ridicate de stres, ci și ca măsură preventivă împotriva incidenței ridicate a avorturilor spontane menționate mai sus. Această formă de glucocorticoid este favorizată datorită metabolizării sale de către enzima placentară 11 Oh steroid dehidrogenază ii, care împiedică glucocorticoidul să aibă vreun efect asupra fătului (71). La momentul concepției, doza de hidrocortizon trebuie crescută la 20-25 mg/zi, iar modificările dozei se efectuează la fiecare 6-8 săptămâni, cu scopul de a menține nivelurile de testosteron la nivelurile normale superioare ale trimestrului de sarcină (74). De exemplu, sunt prezentate valorile testosteronului și D4 ale unui pacient cu NCCAH de la concepție până la 3 ani după livrarea a doi gemeni sănătoși (Figura 1).
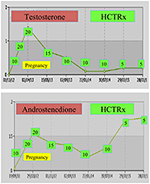
Figura 1. Nivelurile de testosteron și andostenedionă și dozarea ulterioară a hidrocortizonului (HCTRx), de la concepție până la 3 ani după naștere, administrate unui pacient cu NCCAH.
alte probleme speciale
managementul stresului în NCCAH
în timpul stresului major care pune viața în pericol, al intervenției chirurgicale sau al bolilor grave, pacienții cu NCCAH care sunt tratați cu glucocorticoizi pot necesita doze mai mari sau mai frecvente de glucocorticoizi, având în vedere că funcția lor suprarenală este suprimată iatrogenic. Prin urmare, este esențial să educăm părinții copiilor mici, precum și să reeducăm pacienții cu privire la tranziția lor la îngrijirea adulților, cu privire la dozarea stresului. Pentru pacienții cu NCCAH care nu sunt tratați cu GCs sau în cazul întreruperii tratamentului, trebuie evaluat răspunsul cortizolului la ACTH. Aproape o treime dintre pacienții cu NCCAH răspund inadecvat la stimulare (15, 75). Pentru cei care răspund în mod normal la stimularea ACTH nu se recomandă tratament cu doze de stres (19). Terapia cu mineralocorticoizi nu este necesară în niciunul dintre cazuri, deoarece acești pacienți au secreție normală de aldosteron și nu dezvoltă risipă de sare (58). De remarcat, în cazul hipertiroidismului sau al creșterii tratamentului cu levotiroxină, clearance-ul cortizolului este crescut și poate apărea o criză suprarenală (58, 76).
aspecte Psihobiologice în NCCAH
Meyer-Bahlburg și colab. în mai multe studii au raportat afectarea profilului psihologic în rândul pacienților cu NCCAH din cauza deficienței de 21oh. Mai precis, femeile afectate au avut un scor mai mare de masculinizare / defeminizare pe mai multe măsuri de comportament legat de gen în comparație cu femeile normale de control, deși semnificativ mai puțin decât la femeile cu CAH clasic. De asemenea, au crescut semnificativ orientarea bisexuală sau homosexuală. Interviurile clinico-calitative Retrospective cu aceste femei au relevat un istoric de disconfort și stres social legat de experiențele lor de pre-tratament cu semne dependente de androgeni, cum ar fi acneea, hirsutismul și dificultățile de concepție (59, 77, 78). În mod similar, Arlt și colab. a arătat că starea de sănătate subiectivă a fost afectată semnificativ în toate cele opt domenii ale unui sondaj de sănătate pe termen scurt, cu cele mai proeminente diferențe, în comparație cu controalele potrivite vârstei și sexului, legate de domeniile sănătate generală, vitalitate și limitări de rol din cauza problemelor emoționale. Chestionarul Hospital Anxiety and Depression Scale (HADS) a relevat, de asemenea, scoruri crescute de anxietate (41). Având în vedere aceste constatări, parametrii psihologici pentru ghidarea terapiei trebuie luați în considerare la femeile cu NCCAH și, în contextul abordării orientate spre pacient, trebuie oferite un diagnostic psihologic și sprijin.
concluzii
NCCAH este considerată forma mai frecventă și mai ușoară a CAH datorită retenției activității enzimatice de 20-50%. Majoritatea pacienților pot solicita sfatul medicului în orice stadiu al vieții lor din cauza simptomelor legate de excesul de androgeni. Acestea includ pubarche prematură, creștere rapidă, hirsutism, acnee, nereguli menstruale sau infertilitate, precum și multe alte manifestări mai puțin proeminente. Valorile inițiale dimineața devreme de 17 OHP ca un bun test de screening inițial și o evaluare ulterioară cu stimularea ACTH și, în cazul rezultatelor limită, se recomandă testarea genetică. Ca regulă generală, în timpul urmăririi trebuie utilizate niveluri de androstendionă și nu 17 OHP.
decizia de tratament trebuie să se bazeze pe evaluarea faptelor și trebuie să urmeze o abordare individualizată. Tratamentul nu este întotdeauna indicat decât dacă pacientul este simptomatic, de exemplu, copii cu debut precoce și progresie rapidă a părului pubian și corporal, creștere rapidă și/sau avansare scheletică sau femei cu oligomenoree, acnee, hirsutism, infertilitate sau o combinație a acestora și a altora dintre simptomele menționate mai sus. Consilierea genetică este recomandată cu tărie la femeile NCCAH care doresc să conceapă, precum și genotiparea tatălui. Managementul general al pacientului include în plus gestionarea complicațiilor probabile ale terapiei cu glucocorticoizi sau a manifestărilor legate de metabolism ale bolii. În cele din urmă, având în vedere că multe probleme terapeutice legate de gestionarea adecvată a acestor pacienți nu au fost încă elucidate, este foarte important ca medicul curant să fie la curent cu toate evoluțiile din acest domeniu și să integreze noile date în practica sa clinică. Desigur, studii clinice suplimentare în acest domeniu sunt esențiale.
pentru a rezuma, pentru femeia NCCAH, abordarea ideală este una personalizată, încorporând o tranziție lină a managementului ei odată ce este trimisă de la Pediatrie la endocrinologul adult, împreună cu tratamentul orientat spre simptome care o va însoți pe tot parcursul vieții. Având în vedere multiplii factori care afectează sistemul hiperandrogenic, este recomandabil să se încurajeze pacienții să adopte un stil de viață mai sănătos, îmbunătățind obiceiurile alimentare, sporind exercițiile fizice și urmărind reducerea greutății. În plus, în anumite cazuri, sprijinul psihologic este adesea benefic. Mai mult, specialiștii din domeniile implicate în tratamentul acestei tulburări, cum ar fi dermatologii, ginecologii și psihologii, au nevoie de o înțelegere aprofundată cu privire la gestionarea cazurilor suspecte sau deja diagnosticate de NCCAH. În concluzie, să ținem cont de citatul perspicace al astronomului american Vera Cooper Rubin: „știința progresează cel mai bine atunci când observațiile ne obligă să ne modificăm preconcepțiile.”
contribuțiile autorului
toți autorii enumerați au adus o contribuție substanțială, directă și intelectuală la lucrare și au aprobat-o pentru publicare.
Declarație privind conflictul de interese
autorii declară că cercetarea a fost realizată în absența oricăror relații comerciale sau financiare care ar putea fi interpretate ca un potențial conflict de interese.
1. Speiser PW, alb PC. Hiperplazie suprarenală congenitală. N Engl J Med. (2003) 349:776–88. doi: 10.1056/NEJMra021561
PubMed Abstract | CrossRef Full Text/Google Scholar
2. Turcu AF, Auchus RJ. Adrenal steroidogenesis and congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. (2015) 44:275–96. doi: 10.1016/j.ecl.2015.02.002
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Speiser PW. Nonclassic adrenal hyperplasia. Rev Endocr Metab Disord. (2009) 10:77–82. doi: 10.1007/s11154-008-9097-x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Gidl s, Falhammar H, Thil s, von D S, Ritz s, Wedell a și colab. O sută de ani de hiperplazie suprarenală congenitală în Suedia: un studiu de cohortă retrospectiv, bazat pe populație. Lancet Diabet Endocrinol. (2013) 1:35–42. doi: 10.1016/S2213-8587(13)70007-X
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, Noul MI. Frecvență ridicată a deficitului nonclasic de steroizi 21-hidroxilază. Sunt Jum Genet. (1985) 37:650–67.
PubMed Abstract | Google Scholar
6. Wilson RC, Nimkarn S, Dumic M, Obeid J, Azar MR, Azar M, et al. Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Mol Genet Metab. (2007) 90:414–21. doi: 10.1016/j.ymgme.2006.12.005
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Miller WL. Clinical review 54: genetics, diagnosis, and management of 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1994) 78:241–6. doi: 10.1210/jcem.78.2.8106606
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Witchel SF, Azziz R. Nonclassic congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2010) 2010:625105. doi: 10.1186/1687-9856-2010-625105
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Wilson RC, Mercado AB, Cheng KC, noul MI. Deficitul de steroizi 21-hidroxilază: genotipul nu poate prezice fenotipul. J Clin Endocrinol Metab. (1995) 80:2322–9. doi: 10.1210 / jc.80.8.2322
CrossRef Full Text | Google Scholar
10. PC alb. Screening Neonatal pentru hiperplazia suprarenală congenitală. Nat Rev Endocrinol. (2009) 5:490–8. doi: 10.1038/nrendo.2009.148
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Kohn B, Levine LS, Pollack MS, Pang S, Lorenzen F, Levy D, et al. Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. (1982) 55:817–27. doi: 10.1210/jcem-55-5-817
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Moran C, Azziz R, Carmina e, Dewailly D, Fruzzetti F, iba l, și colab. Hiperplazia suprarenală nonclasică cu deficit de 21-hidroxilază este o tulburare progresivă: un studiu multicentric. Am J Obstetrician Ginecol. (2000) 183:1468–74. doi: 10.1067 / gloată.2000.108020
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Azziz R, Hincapie LA, Knochenhauer ES, Dewailly D, Fox L, Cizme LR. Screening pentru hiperplazia suprarenală nonclasică cu deficit de 21-hidroxilază în rândul femeilor hiperandrogenice: un studiu prospectiv. Fertil Steril. (1999) 72:915–25. doi: 10.1016/S0015-0282(99)00383-0
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Livadas S, Dracopoulou M, Dastamani A, Sertedaki A, Maniati-Christidi M, Magiakou a-M, și colab. Spectrul constatărilor clinice, hormonale și moleculare la 280 de persoane cu hiperplazie suprarenală congenitală nonclasică cauzată de mutații ale genei CYP21A2. Clin Endocrinol (Oxf). (2015) 82:543–9. doi: 10.1111 / cen.12543
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Bideu m, Bellann inkt-Chantelot C, Galand-Portier M-B, Tardy V, Billaud l, Laborde K, și colab. Caracterizarea clinică și moleculară a unei cohorte de 161 de femei fără legătură cu hiperplazie suprarenală congenitală nonclasică datorată deficienței de 21-hidroxilază și 330 de membri ai familiei. J Clin Endocrinol Metab. (2009) 94:1570–8. doi: 10.1210 / jc.2008-1582
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Escobar-Morreale HF, Sanch R, San Mill Jl. Un studiu prospectiv al prevalenței hiperplaziei suprarenale congenitale nonclasice în rândul femeilor care prezintă simptome și semne hiperandrogenice. J Clin Endocrinol Metab. (2008) 93:527–33. doi: 10.1210 / jc.2007-2053
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Dewailly D, Vantyghem-Haudiquet MC, Sainsard C, Buvat J, Cappoen JP, Ardaens K, și colab. Fenotipuri clinice și biologice în deficit de 21-hidroxilază cu debut tardiv. J Clin Endocrinol Metab. (1986) 63:418–23. doi: 10.1210 / jcem-63-2-418
PubMed Abstract | CrossRef Full Text/Google Scholar
18. G, oz, Alika, Alika, Alika, Alika, Alikai, Alikai, Alikai, Alikai, Alikai, Alikai, Alikai, Alikai, Alikai, Alikai, Alikai, Alikai, Kandemir N. (2011). Este progesteronul bazal seric 17-OH un parametru fiabil pentru a prezice hiperplazia suprarenală congenitală nonclasică în adrenarche prematură? Turk J Pediatr. 53:274–80.
PubMed Abstract / Google Scholar
19. Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP și colab. Hiperplazia suprarenală congenitală datorată deficienței de steroizi 21-hidroxilază: o orientare clinică a Societății endocrine. J Clin Endocrinol Metab. (2010) 95:4133–60. doi: 10.1210 / jc.2009-2631
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Dacou-Voutetakis C, Dracopoulou M. High incidence of molecular defects of the CYP21 gene in patients with premature adrenarche. J Clin Endocrinol Metab. (1999) 84:1570–4. doi: 10.1210/jc.84.5.1570
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Ambroziak U, Kepczynska-Evenimente I, Kurylowicz, ma Unixtunowicz EM, W Unixtjcicka, Miskiewicz P, și colab. Diagnosticul hiperplaziei congenitale non-clasice a cortexului suprarenale datorită deficienței de 21-hidroxilază, pe baza stimulării serice bazale sau post-ACTH 17-hidroxiprogesteron, poate duce la un diagnostic fals pozitiv. Clin Endocrinol (Oxf). (2016) 84:23–9. doi: 10.1111 / preț.12935
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Ambroziak U, Kepczynska-Evenimente I, Kurylowicz, Wys Xixtouch-cieszy Xixtska A, ma Xixtunowicz EM, Bartoszewicz și colab. LC-MS / MS îmbunătățește screeningul spre deficiența de 21-hidroxilază. Ginecol Endocrinol. (2015) 31:296–300. doi: 10.3109/09513590.2014.994599
PubMed Abstract | CrossRef Full Text | Google Scholar
23. BBC, CC, Goldman MM, Zhang K, Clark-N, Reitz re Welt CK. Măsurarea simultană a treisprezece hormoni steroizi la femeile cu sindromul ovarului polichistic și femeile de control folosind cromatografie lichidă-spectrometrie de masă tandem. PLoS Unu. (2014) 9:e93805. doi: 10.1371/jurnal.pone.0093805
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Ray JA, Kushnir MM, Yost RA, Rockwood AL, Wayne Meikle A. îmbunătățirea performanței în măsurarea a 5 steroizi endogeni prin LC-MS/MS combinat cu spectrometrie de mobilitate ionică diferențială. Clin Chim Acta. (2015) 438:330–6. doi: 10.1016 / j.cca.2014.07.036
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Stoupa A, Gonz Okticlez-Brice l, Pinto G, Samara-Boustani D, Thalassinos C, Flechtner I, și colab. Răspunsul inadecvat al cortizolului la testul tetracosactidei (Synacthen XV) în hiperplazia suprarenală congenitală non-clasică: o excepție de la regulă? Horm Res Pediatru. (2015) 83:262–7. doi: 10.1159/000369901
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. (1992) 90:584–95. doi: 10.1172/JCI115897
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Temeck JW, Pang SY, Nelson C, noul MI. Defecte genetice ale steroidogenezei în pubarche prematură. J Clin Endocrinol Metab. (1987) 64:609–17. doi: 10.1210 / jcem-64-3-609
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Voutilainen R, J Xvskel Xvinen J. Adrenarche prematură: etiologie, constatări clinice și consecințe. J Steroizi Biochem Mol Biol. (2015) 145:226–36. doi: 10.1016/j.jsbmb.2014.06.004
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Oberfield SE, Sopher AB, Gerken AT. Approach to the girl with early onset of pubic hair. J Clin Endocrinol Metab. (2011) 96:1610–22. doi: 10.1210/jc.2011-0225
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Janus D, Wojcik M, Malunowicz E, Starzyk JB. Un caz de aderențe labiale recurente la un copil de 15 luni cu hiperplazie suprarenală congenitală asimptomatică non-clasică din cauza deficienței de 21-hidroxilază. J Pediatr Endocrinol Metab. (2012) 25:1017–21. doi: 10.1515 / jpem-2012-0190
PubMed Abstract | CrossRef Full Text | Google Scholar
31. Gopal-Kothandapani JS, Petkar A, O ‘ Shea E, Banerjee I. părul Perianal ca prezentare neobișnuită a hiperplaziei suprarenale congenitale non-clasice. Cazul BMJ Rep. (2013) 2013:bcr2013010123. doi: 10.1136/bcr-2013-010123
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Falhammar H, Nordenström A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine. (2015) 50:32–50. doi: 10.1007/s12020-015-0656-0
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Chrousos GP, Loriaux DL, Mann DL, Cutler GB. Late-onset 21-hydroxylase deficiency mimicking idiopathic hirsutism or polycystic ovarian disease. Ann Intern Med. (1982) 96:143–8. doi: 10.7326/0003-4819-96-2-143
CrossRef Full Text | Google Scholar
34. Witchel SF. Non-classic congenital adrenal hyperplasia. Steroids. (2013) 78:747–50. doi: 10.1016/j.steroids.2013.04.010
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Carmina E, Rosato F, Jannì A, Rizzo M, Longo RA. Extensive clinical experience: relative prevalence of different androgen excess disorders in 950 women referred because of clinical hyperandrogenism. J Clin Endocrinol Metab. (2006) 91:2–6. doi: 10.1210/jc.2005-1457
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Moran C, Azziz R, Weintrob N, Witchel SF, Rohmer V, Dewailly D, și colab. Rezultatul reproductiv al femeilor cu hiperplazie suprarenală nonclasică cu deficit de 21-hidroxilază. J Clin Endocrinol Metab. (2006) 91:3451–6. doi: 10.1210 / jc.2006-0062
PubMed Abstract | CrossRef Full Text | Google Scholar
37. Levin JH, Carmina E, Lobo RA. Secreția inadecvată de gonadotropină a pacienților cu sindromul ovarului polichistic este similară cu cea a pacienților cu hiperplazie suprarenală congenitală cu debut adult? Fertil Steril. (1991) 56:635–40. doi: 10.1016/S0015-0282(16)54592-0
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Carmina E, Lobo RA. Ovarian suppression reduces clinical and endocrine expression of late-onset congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Fertil Steril. (1994) 62:738–43. doi: 10.1016/S0015-0282(16)56998-2
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Ludwig E. clasificarea tipurilor de alopecie androgenetică (chelie comună) care apar la sexul feminin. Br J Dermatol. (1977) 97:247–54. doi: 10.1111 / j. 1365-2133.1977.tb15179.x
PubMed Abstract | CrossRef Full Text/Google Scholar
40. Trakakis E, Papadavid E, Dalamaga M, Koumaki D, Stavrianeas N, Rigopoulos D, și colab. Prevalența hiperplaziei suprarenale congenitale non-clasice datorată deficienței de 21-hidroxilază la femeile grecești cu acnee: un studiu transversal bazat pe spital. J Eur Acad Dermatol Venereol. (2013) 27:1448–51. doi: 10.1111 / j. 1468-3083.2012. 04613.x
PubMed Abstract | CrossRef Full Text/Google Scholar
41. Arlt W, Willis DS, Wild Sh, Krone N, Doherty EJ, Hahner S, și colab. Starea de sănătate a adulților cu hiperplazie suprarenală congenitală: un studiu de cohortă la 203 pacienți. J Clin Endocrinol Metab. (2010) 95:5110–21. doi: 10.1210 / jc.2010-0917
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Saygili F, Oge a, Yilmaz C. hiperinsulinemia și insensibilitatea la insulină la femeile cu hiperplazie suprarenală congenitală nonclasică datorată deficienței de 21-hidroxilază: relația dintre nivelurile serice de leptină și hiperinsulinemia cronică. Horm Res. (2005) 63:270-4. doi: 10.1159/000086363
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Speiser PW, Serrat J, New MI, Gertner JM. Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1992) 75:1421–4. doi: 10.1210/jc.75.6.1421
PubMed Abstract | CrossRef Full Text | Google Scholar
44. Han TS, Stimson RH, Rees DA, Krone N, Willis DS, Conway GS și colab. Regimul de tratament cu glucocorticoizi și rezultatele de sănătate la adulții cu hiperplazie suprarenală congenitală. Clin Endocrinol. (2013) 78:197–203. doi: 10.1111 / cen.12045
PubMed Abstract | CrossRef Full Text | Google Scholar
45. Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. utilizarea corticosteroizilor orali și riscul de fracturi. J Bone Miner Res. (2000) 15:993-1000. doi: 10.1359 / jbmr.2000.15.6.993
PubMed Abstract | CrossRef Full Text | Google Scholar
46. Nebesio TD, Renbarger JL, Nabhan ZM, Ross SE, Slaven JE, Li L, et al. Differential effects of hydrocortisone, prednisone, and dexamethasone on hormonal and pharmacokinetic profiles: a pilot study in children with congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2016) 2016:17. doi: 10.1186 / s13633-016-0035-5
PubMed Abstract | CrossRef Full Text/Google Scholar
47. Weintrob n, Dickerman Z, Sprecher e, Galatzer A, Pertzelan A. deficiență Non-clasică de 21-hidroxilază în copilărie și copilărie: efectul timpului de inițiere a terapiei asupra pubertății și înălțimii finale. EUR J Endocrinol. (1997) 136:188–95. doi: 10.1530 / eje.0.1360188
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Bonfig W, Bechtold S, Schmidt H, Knorr D, Schwarz HP. Reduced final height outcome in congenital adrenal hyperplasia under prednisone treatment: deceleration of growth velocity during puberty. J Clin Endocrinol Metab. (2007) 92:1635–9. doi: 10.1210/jc.2006-2109
PubMed Abstract | CrossRef Full Text | Google Scholar
49. Dörr HG. Creșterea la pacienții cu hiperplazie suprarenală congenitală clasică datorată deficienței de 21-hidroxilază. Horm Res. (2007) 68 (Supliment 5): 93-9. doi: 10.1159 / 000110587
PubMed Abstract | CrossRef Full Text | Google Scholar
50. Hoepffner W, Kaufhold A, Willgerodt H, Keller E. pacienții cu hiperplazie suprarenală congenitală clasică datorată deficienței de 21-hidroxilază își pot atinge înălțimea țintă: experiența de la Leipzig. Horm Res. (2008) 70:42-50. doi: 10.1159/000129677
PubMed Abstract | CrossRef Full Text | Google Scholar
51. van der Kaay D, van den Akker E. Ultralow-dose dexamethasone to preserve endogenous cortisol stress response in nonclassical congenital adrenal hyperplasia: a new promising treatment. Int J Endocrinol Metab. (2014) 12:e14657. doi: 10.5812/ijem.14657
CrossRef Full Text | Google Scholar
52. Lin-Su K, Vogiatzi MG, Marshall I, Harbison MD, Macapagal MC, Betensky B, et al. Treatment with growth hormone and luteinizing hormone releasing hormone analog improves final adult height in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. (2005) 90:3318–25. doi: 10.1210/jc.2004-2128
PubMed Abstract | CrossRef Full Text | Google Scholar
53. McCann-Crosby B, Chen M-J, Lyons SK, Lin Y, Axelrad M, Dietrich JE, et al. Nonclassical congenital adrenal hyperplasia: obiective de tratament și tranziție. Pediatrul Endocrinol Rev. (2014) 12:224-38.
PubMed Abstract | Google Scholar
54. Ogilvie CM, Crouch NS, Rumsby G, Creighton SM, Liao L-M, Conway GS. Hiperplazia suprarenală congenitală la adulți: o revizuire a problemelor medicale, chirurgicale și psihologice. Clin Endocrinol. (2006) 64:2–11. doi: 10.1111 / j. 1365-2265.2005. 02410.x
PubMed Abstract | CrossRef Full Text | Google Scholar
55. Mah PM, Jenkins RC, Rostami-Hodjegan A, Newell-Price J, Doane A, Ibbotson V, et al. Weight-related dosing, timing and monitoring hydrocortisone replacement therapy in patients with adrenal insufficiency. Clin Endocrinol. (2004) 61:367–75. doi: 10.1111/j.1365-2265.2004.02106.x
PubMed Abstract | CrossRef Full Text | Google Scholar
56. Diaz A, Laufer dl pelviana LLAA de PC Pe A, îngrijire AC de O și GC pe AH. Menstruația la fete și adolescenți: utilizarea ciclului menstrual ca semn vital. Pediatrie. (2006) 118:2245–50. doi: 10.1542 / peds.2006-2481
CrossRef Full Text | Google Scholar
57. Merke DP, Poppas DP. Tratamentul adolescenților cu hiperplazie suprarenală congenitală. lancet diabet Endocrinol. (2013) 1:341–52. doi: 10.1016/S2213-8587(13)70138-4
PubMed Abstract | CrossRef Full Text | Google Scholar
58. Verma S, Green-Golan L, VanRyzin C, Drinkard B, Mehta SP, Weise M, et al. Adrenomedullary function in patients with nonclassic congenital adrenal hyperplasia. Horm Metab Res. (2010) 42:607–12. doi: 10.1055/s-0030-1253385
PubMed Abstract | CrossRef Full Text | Google Scholar
59. New MI. Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2006) 91:4205–14. doi: 10.1210/jc.2006-1645
PubMed Abstract | CrossRef Full Text | Google Scholar
60. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2018) 103:4043–88. doi: 10.1210/jc.2018-01865
PubMed Abstract | CrossRef Full Text | Google Scholar
61. Kruse B, Riepe FG, Krone N, Bosinski HAG, Kloehn S, Partsch CJ, et al. Congenital adrenal hyperplasia – how to improve the transition from adolescence to adult life. Exp Clin Endocrinol Diabetes. (2004) 112:343–55. doi: 10.1055/s-2004-821013
PubMed Abstract | CrossRef Full Text | Google Scholar
62. Gastaud F, Bouvattier C, Duranteau L, Brauner R, Thibaud e, Kutten F, și colab. Afectarea rezultatelor sexuale și reproductive la femeile cu forme clasice de hiperplazie suprarenală congenitală. J Clin Endocrinol Metab. (2007) 92:1391–6. doi: 10.1210 / jc.2006-1757
PubMed Abstract | CrossRef Full Text | Google Scholar
63. Merc Fern inktiftndez-Balsells m, Muthusamy K, Smushkin G, Lampropulos JF, ELAMIN MB, Abu Elnour NO, și colab. Utilizarea prenatală a dexametazonei pentru prevenirea virilizării la sarcini cu risc de hiperplazie suprarenală congenitală clasică din cauza deficienței 21-hidroxilazei (CYP21A2): o revizuire sistematică și meta-analize. Clin Endocrinol. (2010) 73:436–44. doi: 10.1111 / j. 1365-2265.2010. 03826.x
CrossRef Full Text/Google Scholar
64. Bideul m, Bellann-Chantelot c, Galand-Portier MB, Golmard JL, Tardy V, Morel Y și colab. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2010) 95:1182–90. doi: 10.1210/jc.2009-1383
PubMed Abstract | CrossRef Full Text | Google Scholar
65. Birnbaum MD, Rose LI. Late onset adrenocortical hydroxylase deficiencies associated with menstrual dysfunction. Obstet Gynecol. (1984) 63:445–51.
PubMed Abstract | Google Scholar
66. Cuhaci N, Aydin C, Yesilyurt A, Pinarli FA, Ersoy R, Cakir B. Nonclassical congenital adrenal hyperplasia and pregnancy. Case Rep Endocrinol. (2015) 2015:296924. doi: 10.1155/2015/296924
PubMed Abstract | CrossRef Full Text | Google Scholar
67. Deneux C, Tardy V, Anne D, Mornet E, Billaud L, Charron D, et al. Corelația fenotip-genotip la 56 de femei cu hiperplazie suprarenală congenitală nonclasică datorată deficitului de 21-hidroxilază. J Clin Endocrinol Metab. (2001) 86:207–13. doi: 10.1210 / jcem.86.1.7131
PubMed Abstract | CrossRef Full Text | Google Scholar
68. Simonetti L, BRUQUE CD, Fern Inktivndez CS, Benavides-Mori B, Delea M, Kolomenski JE și colab. Actualizarea mutației CYP21A2: analiza cuprinzătoare a bazelor de date și a variantelor genetice publicate. Hum Mutat. (2018) 39:5–22. doi: 10.1002 / humu.23351
PubMed Abstract | CrossRef Full Text | Google Scholar
69. OMIM Entry – * 613815 – CYTOCHROME P450 FAMILY 21 SUBFAMILY A POLYPEPTIDE 2; CYP21A2.
Google Scholar
70. Miller WL, Witchel SF. Prenatal treatment of congenital adrenal hyperplasia: risks outweigh benefits. Am J Obstet Gynecol. (2013) 208:354–9. doi: 10.1016/j.ajog.2012.10.885
PubMed Abstract | CrossRef Full Text | Google Scholar
71. Lekarev O, New MI. Adrenal disease in pregnancy. Best Pract Res Clin Endocrinol Metab. (2011) 25:959–73. doi: 10.1016/j.beem.2011.08.004
PubMed Abstract | CrossRef Full Text | Google Scholar
72. New MI, Abraham M, Yuen T, Lekarev O. An update on prenatal diagnosis and treatment of congenital adrenal hyperplasia. Semin Reprod Med. (2012) 30:396–9. doi: 10.1055/s-0032-1324723
PubMed Abstract | CrossRef Full Text | Google Scholar
73. Kazmi D, Bailey J, Yau M, Abu-Amer W, Kumar A, Low M, et al. New developments in prenatal diagnosis of congenital adrenal hyperplasia. J Steroid Biochem Mol Biol. (2017) 165:121–3. doi: 10.1016/j.jsbmb.2016.06.016
PubMed Abstract | CrossRef Full Text | Google Scholar
74. Clayton PE, Miller WL, Oberfield SE, Ritz, Sippell WG, Speiser PW și colab. Declarație de consens privind deficiența de 21-hidroxilază din partea Societății Europene de endocrinologie pediatrică și a Societății endocrine pediatrice Lawson wilkins. Horm Res. (2002) 58:188-95. doi: 10.1159 / 000065490
PubMed Abstract | CrossRef Full Text | Google Scholar
75. Nandagopal R, Sinaii N, Avila NA, Van Ryzin C, Chen W, Finkielstain GP, și colab. Profilarea fenotipică a părinților cu hiperplazie suprarenală congenitală criptică nonclasică: constatări la 145 de familii fără legătură. EUR J Endocrinol. (2011) 164:977–84. doi: 10.1530 / EJE-11-0019
PubMed Abstract | CrossRef Full Text | Google Scholar
76. Takasu n, Nakachi K, Higa H. dezvoltarea hipertiroidismului Graves a provocat o criză suprarenală la un pacient cu deficiență de 21-hidroxilază non-clasică nerecunoscută anterior. Intern Med. (2010) 49:1395–400. doi: 10.2169 / internalmedicine.49.3573
PubMed Abstract | CrossRef Full Text | Google Scholar
77. Meyer-Bahlburg HFL, Dolezal C, Baker SW, Carlson AD, Obeid JS, New MI. Prenatal androgenization affects gender-related behavior but not gender identity in 5-12-year-old girls with congenital adrenal hyperplasia. Arch Sex Behav. (2004) 33:97–104. doi: 10.1023/B:ASEB.0000014324.25718.51
CrossRef Full Text | Google Scholar
78. Meyer-Bahlburg HFL, Dolezal C, Baker SW, noul MI. Orientarea sexuală la femeile cu hiperplazie suprarenală congenitală clasică sau non-clasică în funcție de gradul de exces de androgeni prenatali. Arc Sex Se Comporte. (2008) 37:85–99. doi: 10.1007 / s10508-007-9265-1
PubMed Abstract / CrossRef Full Text / Google Scholar