a estrutura primária de uma proteína é definida como a sequência de aminoácidos de que é composta. Esta sequência finalmente determina a forma que a proteína adota, de acordo com as limitações espaciais sobre a disposição dos átomos na proteína, as propriedades químicas do componente resíduos de aminoácidos, e o ambiente da proteína.as ligações peptídicas que ligam os resíduos de aminoácidos num polipéptido formam-se numa reacção de condensação entre o grupo carboxilo ácido de um aminoácido e o grupo amino básico de outro aminoácido. No contexto de um peptídeo, o grupo amida (CO–NH) é referido como o grupo peptídeo.
Crucial para a compreensão da estrutura proteica é o conhecimento da estrutura da ligação peptídica. Linus Pauling, in the 1930s, used X-ray diffraction to examine the nature of the peptide bond formed between two amino acids. He reported that the peptide group (CO–NH) has a rigid planar structure. Esta estrutura é devida a interações entre elétrons da dupla ligação do grupo carbonila e aqueles da ligação C–N (Figura 2) de tal forma que este último adquire propriedades parciais (cerca de 40%) de dupla ligação.

este efeito é um exemplo de ressonância que pode ser pensado como uma partilha de elétrons entre ligações. Uma vez que as ligações únicas entre dois átomos são mais longas que as ligações duplas entre os mesmos dois átomos, os comprimentos das ligações C–N E C=o no grupo peptídeo diferem dos observados para essas ligações em outros contextos onde a ressonância não ocorre. Assim, o caráter parcial de ligação dupla de C-N no grupo peptídico significa que esta ligação é mais curta do que seria previsto para uma ligação única C–N, enquanto a ligação C=o, tendo um caráter parcial de ligação única devido à ressonância, é maior do que seria previsto para uma ligação dupla C=O. Os comprimentos das ligações no grupo peptídeo estão indicados na Figura 3. Compare a ligação C–N do grupo peptídico com a ligação entre N e Ca (o átomo C ao qual o grupo amino e o grupo carboxilo estão ligados).

Existem duas possíveis conformações da ligação péptida planar: no grupo do peptídeo trans, os átomos de Ca estão em lados opostos da ligação peptídica (figura 3a) e no grupo do peptídeo cis, os átomos de Ca estão do mesmo lado da ligação peptídica (figura 3b).
-
considerando a disposição espacial e a proximidade dos átomos na cis e as trans conformações da ligação peptídica, que conformação você acha que seria favorecida?
-
a trans conformação seria energeticamente mais favorável do que a conformação do SCI, uma vez que minimiza o obstáculo estérico.
de um modo geral, as ligações peptídicas estão na conformação trans. No entanto, as formas cis podem ocorrer em ligações peptídicas que precedem um resíduo prolina. Nesses casos, a forma cis é mais estável do que o habitual, uma vez que a cadeia lateral prolina oferece menos obstáculos. No entanto, as ligações com peptídeo cis ocorrem apenas em aproximadamente 10% dos casos de ligações peptídicas anteriores aos resíduos da prolina.tendo em conta a natureza planar do grupo peptídeo, uma cadeia polipeptídica pode ser vista como tendo uma espinha dorsal que consiste numa série de grupos rígidos de peptídeo planar ligados pelos átomos da Ca. A figura 4 mostra parte de um polipéptido com dois grupos peptídicos planares na conformação trans. Note que embora a rotação não seja permitida sobre as ligações peptídicas, há potencial para rotação em torno das ligações Ca–N e Ca–C. Os ângulos de rotação, denominados ângulos de torção , sobre estas ligações especificam a conformação de uma espinha dorsal polipéptida. Os ângulos de torção sobre as ligações Ca–n e Ca-C são referidos como ɸ (phi) e ψ. (psi), respectivamente, e são definidos como 180° quando o polipéptido está na conformação planar estendida, como ilustrado na Figura 4.
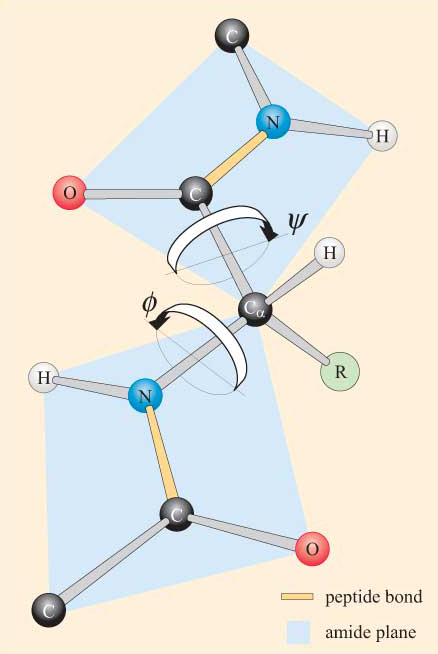
não ficará surpreendido ao saber que as restrições estéricas se aplicam a ψ e ψ.
como resultado destas restrições estéricas, apenas certos valores de ψ e ψ, e portanto conformações do péptido, são permitidos enquanto outros não são.
é possível calcular estes valores permitidos para um determinado resíduo no contexto de um polipeptídeo. Este cálculo é realizado primeiro determinando as distâncias entre todos os átomos não ligados em dois grupos peptídicos vizinhos (como os da Figura 4) em todos os valores possíveis de ψ e ψ. É mais facilmente feito para um polipéptido contendo apenas um tipo de aminoácido. A conformational plot of ψ against ψ for a particular residue is known as a Ramachandran plot (after its inventor, G. N. Ramachandran). Tal gráfico permite-nos identificar as conformações (isto é, para um valor particular de ψ e ψ) Que são estericamente favoráveis ou desfavoráveis (como Na Figura 5), de acordo com os seguintes critérios:
-
Onde não há conflito entre os raios de van der Waals de átomos não ligados, uma conformação é “permitida”. Estas conformações encontram-se nas áreas azuis da Figura 5.
-
As conformações que exigem distâncias interatómicas ao limite daquilo que é admissível são definidas como conformações de “limite exterior”. Encontram-se nas zonas verdes da Figura 5.
-
conformações teóricas que exigem que quaisquer dois átomos não-ligantes estejam mais próximos um do outro do que os seus raios de van der Waals permitem são estericamente “proibidos”. Estes encontram-se nas áreas brancas da Figura 5.
Notice that the values of ɸ and ψ in Figure 5 range from-180 ° to + 180°. Rodar o grupo peptídeo até 360º irá, naturalmente, trazê −lo de volta à sua posição inicial, e-180º e +180º correspondem à mesma posição. Assim, a faixa verde no canto inferior esquerdo da parcela na Figura 5 é contígua com o campo no canto superior esquerdo.

-
Utilizar a Figura 5 para determinar se os seguintes valores de ɸ e ψ são sterically favoráveis ou desfavoráveis: (a) ɸ = 90º e ψ = 90º; (b) ɸ = −90º e ψ = 90º.
-
prolina também é bastante diferente de outros aminoácidos em termos de conformações Permitidas e para poliprolina apenas são tolerados valores de −85º a −35º. Pensando sobre a estrutura da prolina, como você pode explicar esta faixa relativamente estreita de valores autorizados ɸ?
-
a cadeia lateral da prolina está ligada covalentemente ao N do grupo amino, pelo que na poliprolina haverá menos liberdade de rotação sobre a ligação Ca–N do que com outros aminoácidos. Consequentemente, os valores de ɸ permitidos serão relativamente limitados em comparação com outros aminoácidos.
-
A Figura 6 mostra a parcela Ramachandran de resíduos de glicina numa cadeia polipeptídica. As regiões são codificadas por cores, como indicado na Figura 5. O que você pode dizer sobre as conformações que a glicina adota? Considere a estrutura da glicina. Por que a glicina difere dos outros resíduos no que diz respeito às suas conformações?
-
glicina tem uma liberdade de conformação muito maior do que os outros resíduos de aminoácidos, porque é menos estericamente dificultada.
-
olhe novamente para a Figura 4 e imagine que pode torcer o grupo peptídeo mais alto através de 180° de modo que ψ = 0. Que grupos são susceptíveis de entrar em conflito nesta conformação?
-
os grupos N–H de grupos péptidos adjacentes entram em conflito uns com os outros, sendo forçados a aproximarem-se.
(a) desfavorável; B) favorável.as parcelas de Ramachandran podem ser construídas para polímeros de cada um dos 20 aminoácidos. É significativo notar que as parcelas de Ramachandran para muitos resíduos de aminoácidos são geralmente muito semelhantes, tendo apenas três regiões com conformações favoráveis ou toleradas (1-3 na parcela para poli-L-alanina na Figura 5). No entanto, ocorrem diferenças. Por exemplo, onde a cadeia lateral (R Na Figura 4) é ramificada perto de Ca, como no caso da treonina, ocupa mais espaço perto da espinha dorsal do péptido e restringe a aproximação dos átomos nos grupos peptídicos vizinhos. Como resultado, conformações permitidas (ɸ e ψ ângulos) são mais restritas para polipéptidos de aminoácidos ramificados.

O Ramachandran parcelas nas Figuras 5 e 6 foram gerados, respectivamente, l-alanina e l-glicina na base do permitido e o limite exterior distâncias para interatomic contatos, determinada a partir de valores conhecidos de van der Waals raios dos átomos (Tabela 1).
Table 1 Van der Waals distances for interatomic contacts.
| tipo de Contato | Normalmente permitidos / Å | limite Exterior / Å | |
|---|---|---|---|
| H···H | 2.0 | 1.9 |
3.0 |
são, portanto, gráficos preditivos e não conformacionais reais. Nós podemos, naturalmente, usar difração de raios X para determinar experimentalmente os valores ‘reais’ de ɸ e ψ Para Resíduos em um polipeptídeo. Na Figura 7, Os valores de ψ e ψ Para todos os resíduos (com exceção de glicina e prolina) em uma série de estruturas diferentes foram determinados por difração de raios X de alta resolução e plotados em uma parcela Ramachandran. Podemos ver que existe uma correspondência impressionante entre as conformações previstas e reais. Note, no entanto, que existem alguns resíduos cujas conformações mapeiam as áreas “proibidas”. A maioria destes resíduos mapeiam na região entre as regiões’ permitidas ‘ 2 e 3, em torno de ψ = 0.

o conflito associado a estas conformações pode ser acomodado por um pequeno grau de torção da ligação peptídica. Assim, em tais conformações o grupo peptídico é torcido para fora de sua conformação planar usual.um número limitado de conformações “proibidas” de determinados resíduos pode ser tolerado num polipéptido se a conformação adoptada, no seu conjunto, for energeticamente favorável. Um polipéptido tende a dobrar de tal forma que adota a conformação mais estável. Nesta conformação, o polipéptido minimiza a sua energia livre. Nas secções seguintes, analisaremos este nível mais elevado de estrutura proteica.