- definities en prevalentie
- diagnose
- NCCA manifestaties bij vrouwen
- manifestaties in de kindertijd
- manifestaties in de adolescentie en het volwassen leven
- NCCA-behandeling vanaf de eerste manifestatie tot het volwassen leven
- behandeling tijdens de kindertijd
- behandeling tijdens de adolescentie
- patiënten die sinds de kindertijd worden behandeld
- vrouwen met de eerste manifestatie tijdens de adolescentie of Hyperandrogene symptomen na stopzetting van de behandeling
- NCCA en reproductie
- subfertiliteit
- miskramen
- Fertiliteitsplanning
- behandeling tijdens de zwangerschap
- andere speciale problemen
- stressmanagement bij NCCAH
- Psychobiologische aspecten in NCCAH
- conclusies
- Auteursbijdragen
- belangenconflict verklaring
definities en prevalentie
congenitale bijnierhyperplasie (CAH) omvat een familie van autosomaal recessieve stoornissen gekarakteriseerd door lichte tot acuut gestoorde cortisolsynthese als gevolg van een deficiëntie in een van de vijf bijniersteroidogene enzymen die nodig zijn voor de cortisolproductie (1, 2). Conventioneel, CAH is onderverdeeld in (a) klassiek (CCAH), presenteren met zout-wasting of de eenvoudige virilizing vorm die zich manifesteert bij de geboorte en/of in de neonatale periode, en (b) niet-klassiek (NCCAH), die een minder ernstige vorm van de aandoening die genitale ambiguïteit ontbreekt, is niet onmiddellijk levensbedreigend, en presenteert later in het leven, of blijft asymptomatisch, of wordt verkeerd gediagnosticeerd als een andere ziekte (3). Nochtans, zijn de grenzen over de verschillende vormen van CAH niet strikt gedefinieerd, die de neiging heeft om de uitdagingen verbonden aan deze wanorde te verhogen. Het is daarom raadzaam CAH te beschouwen als een continuüm van fenotypen, van ernstig tot mild of anders asymptomatisch (3).
de meest voorkomende vorm van CAH wordt veroorzaakt door 21-hydroxylasedeficiëntie (21OHD), wat resulteert in een verminderde of geen omzetting van 17 hydroxyprogesteron (17 OHP) naar 11 deoxycortisol en van progesteron naar deoxycorticosteron. De blokkade van steroid omzetting resulteert in verhoogde productie van androgen voorlopers, onder CRH-ACTH stimulatie, die tot biochemisch hyperandrogenism leiden, die door opgeheven 17-OHP niveaus wordt gekenmerkt. De klassieke vorm van de ziekte, komt voor bij 1 van de 16.000 levendgeborenen wereldwijd. Er moet echter worden vermeld dat de waargenomen incidentie van de ziekte gerelateerd is aan de gebruikte screeningsmethode. Er is bijvoorbeeld gemeld dat de incidentie van de klassieke vorm van CAH in Zweden 1:11.500 is wanneer een case survey benadering wordt gebruikt, die echter daalt tot 1:9.800 wanneer hormonale screening wordt toegepast. Evenzo varieert de incidentie in Frankrijk en Zwitserland tussen 1:15.472 en 1:23.000 en 1:10.749 en 1:11.661 wanneer de verschillende screeningsmethoden worden gebruikt (4).
NCCAH komt veel vaker voor, komt voor bij ongeveer 1 op de 1.000 Kaukasiërs en vaker bij bepaalde etnische groepen, zoals Asjkenazische Joden (1:27), Hispanics (1:53), Joegoslaven (1: 62) en Italianen (1:300) (5, 6). Niettemin ontbreken gegevens over de prevalentie van NCCAH op basis van verschillende schattingsmethoden (case-survey, hormonale screening en moleculaire testen). NCCAH wordt beschouwd als de meest voorkomende autosomaal recessieve endocriene stoornis met een draagfrequentie van 1:25 tot 1:10. Bij NCCA als gevolg van 21-OHD wordt de resterende enzymatische activiteit geschat op ongeveer 10-70% (7-9).
diagnose
in vergelijking met de diagnose van de klassieke vorm van de ziekte, die wordt gesteld bij de geboorte of tijdens de neonatale periode vanwege genitale ambiguïteit en/of zoutverslindende symptomen of door middel van screeningsprogramma ‘ s die in sommige landen worden toegepast, zijn de meeste gevallen van NCCAH niet gemakkelijk op te sporen (4, 10). Bovendien, veel individuen blijven asymptomatisch tijdens de kindertijd en adolescentie, hebben een normale reproductieve functie, en pas bewust van NCCAH als gevolg van de diagnose van een ander familielid en de daaruit voortvloeiende testen (11). Echter, de meeste vrouwen met NCCAH zoeken medische hulp wanneer ze symptomen van androgeen overmaat ervaren en, wanneer klinische verdenking vraagt testen, verhoogde basale 17 OHP niveaus zal meer waarschijnlijk dan niet wijzen op een diagnose van NCCAH.
De door de endocriene samenleving voorgestelde klinische richtlijnen bevelen een niet-gestimuleerde uitgangswaarde van 17 OHP aan als screeningsinstrument voor NCCA. Ochtend-17 OHP-spiegels >6 nmol/L in de folliculaire fase bij menstruerende vrouwen moeten verdere evaluatie mogelijk maken, aangezien is aangetoond dat concentraties boven 6 nmol/L 90% van de nccah-individuen vangen (12, 13). Willekeurige metingen van 17 OHP zijn niet nuttig gebleken, omdat deze vaak leveren normale niveaus bij patiënten met NCCAH, en ze zijn bovendien extreem hoog in de luteale fase bij de helft van gezonde vrouwen (13). Echter, in onze gegevens afgeleid van 280 proefpersonen met de ziekte, zes patiënten (2.1%) had een baseline 17 OHP-waarde < 6 nmol / L (14). Bovendien, Bidet et al., in een grote cohort van vrouwen met NCCAH geverifieerd door moleculaire technieken vond ook basale 17 OHP waarden lager dan 6 nmol/L bij 8% van de onderzochte proefpersonen (15). Tot slot, gebaseerd op gegevens verzameld door Speiser et al. 9% van de personen met NCCAH vertoonde 17 OHP waarden lager dan 2 ng/ml (dat komt overeen met 6nmol/L). Volgens andere studies is een uitgangswaarde van 17 OHP tussen 5,1 en 9 nmol/L voldoende voor de diagnose van NCCAH (13, 16, 17). Onlangs, een niveau van basaal 17 OHP van 4.6 nmol / L werd voorgesteld als drempelwaarde voor ACTH-testen om NCCAH te voorspellen bij proefpersonen met premature adrenarche tijdens de kindertijd (18).
het totaal van deze bevindingen en suggesties geeft aan dat de selectie van patiënten die een synacthen stimulatietest zullen ondergaan, van geval tot geval moet worden geëvalueerd. Een poststimulatieniveau van 17 OHP boven 3 nmol / L is vereist voor de diagnose (19). Heterozygoten die één CYP21A2-mutatie dragen vertonen licht verhoogde 17 OHP-spiegels na ACTH-stimulatie, hoewel er overlap is bij onaangetaste proefpersonen (9). Echter, als Dacou-Voutetakis et al. hebben erop gewezen, als de som van basale en post-stimulatie 17 OHP waarden hoger is dan 1,5 nmol / L, dan is de mogelijkheid van heterozygositeit uitzonderlijk hoog (20).
een andere belangrijke overweging betreft de technieken die worden gebruikt voor de evaluatie van 17 OHP. 17 OHP niveaus worden gemeten door een verscheidenheid van immunoassay methoden, maar zoals onlangs is aangetoond, werden de meest nauwkeurige en betrouwbare resultaten bereikt door de implementatie van de combinatie van vloeistofchromatografie met massaspectrometrie (LC-MS/MS). Bij vergelijking van LC-MS/Ms-metingen met standaardmethoden werden inderdaad veel vals-positieve 17 OHP-metingen gevonden (21). deze laatste procedures worden echter niet universeel gebruikt, omdat ze in geval van een onzekere diagnose nauwkeurigere resultaten zullen opleveren. De screening-en diagnostische drempels voor LC-MS/MS, gekwantificeerd met 17 OHP, moeten nog worden vastgesteld. Deze drempels zijn waarschijnlijk lager dan die welke zijn vastgesteld met immunoassays vanwege de verhoogde specificiteit van LC-MS/MS, een methode die minder gevoelig is voor kruisreactiviteit en interferenties (22, 23). Of een urinair steroïdprofiel nodig is voor de definitieve diagnose, moet nog worden opgehelderd (22). In borderline gevallen is het raadzaam om na de ACTH stimulatietest een compleet adrenocorticaal profiel te verkrijgen om 21-hydroxylase deficiëntie te onderscheiden van andere enzymdefecten en een stevige diagnose te stellen.
specifiek moet in twijfelachtige gevallen een volledig steroïdprofiel worden uitgevoerd om 21-hydroxylasedeficiëntie te bevestigen en andere enzymatische detecties uit te sluiten. Het opnemen van 17 OHP, cortisol,11-deoxycorticosteron,11-deoxycortisol, 17-OH-pregnenolon, dehydroepiandrosteron en androstenedione in een steroïdprofiel van het serum zou nuttig zijn om andere oorzaken van CAH uit te sluiten, zoals 11β-hydroxylasedeficiëntie en, in zeldzame gevallen, 3β-hydroxysteroïde dehydrogenasedeficiëntie of P450-oxidoreductase-deficiëntie (24). Hoewel genetische tests niet wordt beschouwd als een primaire diagnostische tool voor NCCAH op dit moment, is het verplicht voor diagnose bevestiging en voor genetische counseling (3).
wat de cortisolspiegels betreft, wordt over het algemeen een post-stimulatiewaarde van ten minste 496 nmol/L verwacht. Van nota, volgens Stoupa et al. 60% van de 47 kinderen met NCCAH als gevolg van 21 OHD hadden lage cortisolwaarden na de stimulatie, een bevinding die wijst op de noodzaak van verhoogde surveillance voor de ontwikkeling van bijnierschorsinsufficiëntie tijdens belangrijke stressorgebeurtenissen (25).
NCCA manifestaties bij vrouwen
de klinische expressie van NCCA wordt gekarakteriseerd door een hoog niveau van polymorfisme, niet alleen wat betreft de aanvangsleeftijd, maar ook wat betreft de verschillende tekenen en symptomen. Het is gemeld dat de eerste klinische presentatie van NCCAH is in 11% van de gevallen vóór de leeftijd van 10 jaar en in 80% tussen de leeftijd van 10 en 40 jaar (12). De genotype-fenotype correlatie in CAH en NCCAH is nog niet opgehelderd. Speiser et al. suggereert dat de meeste maar niet alle fenotypische variabiliteit in 21-hydroxylasedeficiëntie het gevolg is van allelische variatie in CYP21A2 (26).
manifestaties in de kindertijd
tijdens de pasgeborene blijven meestal NCCA-vrouwtjes asymptomatisch en hebben normale uitwendige genitaliën. Het vroegste geval van NCCAH gemeld is een 6 maanden oud meisje dat schaamhaar ontwikkeld (11). Meestal, klinische bevindingen en symptomen in NCCAH gevallen beginnen vanaf de leeftijd van 5 jaar of zelfs later en zijn gerelateerd aan verhoogde androgeenspiegels, hoewel milde cortisol deficiëntie kan ook optreden in sommige gevallen (8). In 92% van de nccah-gevallen is het eerste symptoom dat zich manifesteert premature pubarche (PP), die voorkomt onder de 8 jaar bij meisjes en onder de 9 jaar bij jongens (12) met een geschatte prevalentie tussen 10 en 11,3% (15). Van belang is dat bij kinderen met PP, volgens verschillende studies, de incidentie van NCCA varieert van 5 tot 30% (20, 27). Echter, gebaseerd op onze Analyse van 280 patiënten met moleculaire bevestiging van NCCAH, vonden we dat de incidentie van PP bij 94 vrouwen jonger dan 8 jaar was maar liefst 88%. Aan de andere kant identificeerde een analyse van 45 mannen met NCCAH PP bij slechts 29% van de proefpersonen (14). Verder moeten we in gedachten houden dat ongeveer de helft van de personen met PP heterozygote dragers van een CYP21A2 mutatie kan zijn.
een ander aspect dat verduidelijking behoeft is de relatie tussen PP en vroegrijpe puberteit. Er wordt verondersteld dat in sommige gevallen, de perifere aromatisatie van bijnier androgenen aan oestrogenen de hypothalamus-hypofyse-gonadale as kan activeren, leidend tot secundaire vroegrijpe puberteit (28). In dat geval is een regelmatige follow-up nodig. Sommige kinderen ontwikkelen hirsutisme, acne, en / of snelle groei en manifesteren hoge structuur en been leeftijd vooruitgang (29). De laatste kan resulteren in afgeknotte uiteindelijke hoogte als gevolg van snelle epifysaire fusie. Andere zeldzame klinische bevindingen tijdens de kindertijd omvatten labiale adhesie, perianaal haar, clitoromegalie, en heesheid van de stem (30-32).
manifestaties in de adolescentie en het volwassen leven
tijdens de adolescentie en volwassenheid vertoont NCCAH bij vrouwen hyperandrogene symptomen die lijken op polycysteus ovariumsyndroom (PCOS) (33). De symptomen omvatten hirsutism, acne, androgene alopecia, anovulation, menstruele dysfunctie, en onvruchtbaarheid. In een multicenter studie waren de meest voorkomende symptomen bij adolescente en volwassen vrouwen hirsutisme (59%), oligomenorroe (54%) en acne (33%). Bij 161 vrouwen met NCCAH waren de symptomen hirsutisme (78%), menstruatiestoornissen (54,7%) en verminderde vruchtbaarheid (12%) (34). In onze groep van 161 vrouwen met de ziekte had 63% van de patiënten een polycysteus ovarium-achtig fenotype (14). Deze bevinding heeft een vice versa implicatie, omdat de incidentie van NCCA hoog is bij vrouwen met hyperandrogenemie. Zeker, de studie van Witcel et al. het melden van een incidentie van NCCAH variërend van 1 tot 33% bij hyperandrogene vrouwen is van groot belang. In een andere studie van 950 vrouwen die voor evaluatie van androgen overmaat werden verwezen, werd NCCAH ontdekt in 4,2% van hen. Echter, de nccah patiënten waren jonger en meer hirsute in vergelijking met de andere groepen van hyperandrogene patiënten. Ze werden ook gekenmerkt door aanzienlijk hogere niveaus van testosteron, vrij testosteron en 17 OHP dan patiënten met andere hyperandrogene syndromen. In ovariale echografie, 77% van hen vertoond polycystische eierstokken en 41% verhoogde ovariale grootte. In totaal voldeden zij volgens NIH en Rotterdam aan de PCOS-criteria met een percentage van respectievelijk 56 en 72,8% (35).
de progressieve aard van de ziekte wordt benadrukt door het feit dat de prevalentie van hirsutisme is aangetoond te stijgen met de leeftijd en is waargenomen zeldzaam voor de puberteit. Hirsutisme is de meest voorkomende klinische manifestatie gemeld bij patiënten met NCCA, variërend van 71 tot 96% (28, 36). Puberale meisjes met NCCA meestal aanwezig met hirsutisme (37, 38). Aan de andere kant van het spectrum is de kwestie van alopecia. Mannelijke patroon kalende wordt gemeld bij NCCAH patiënten. De prevalentie van alopecia bleek ook te stijgen met de leeftijd, van 6% bij patiënten tijdens het tweede decennium van hun leven tot 19% in het vijfde, wat weer wijst op de progressieve aard van de ziekte (39). Acne wordt gemeld bij bijna 33% van de nccah-proefpersonen (36). Opmerkelijk, werden veranderingen van het CYP21A2 gen die NCCAH veroorzaken ontdekt in 4,9% van 123 volwassen wijfjes die met strenge acne voorstellen (40).
menstruele onregelmatigheden, waaronder oligomenorroe of secundaire amenorroe, kunnen vaak het zichtbare teken zijn van NCCA bij postmenarche individuen. Bovendien ervaren 8-9% van de gevallen ook primaire amenorroe en, om deze reden, voor het eerst medisch advies inwinnen. Bovendien, onder nccah individuen, oligomenorroe komt vaker voor dan primaire amenorroe. Bijvoorbeeld, in een serie van Moran et al. bij NCCAH vrouwen in de leeftijd van 10 tot 19 jaar, oligomenorroe was aanwezig in 56% van de gevallen in vergelijking met slechts 9% van de adolescentie met primaire amenorroe (12). Tot slot, zo veel nccah symptomen lijken op die van PCOS dat het niet verwonderlijk is dat NCCAH is nagesynchroniseerd de grote mimicker van PCOS. In ieder geval moet een diagnose van NCCAH worden overwogen tijdens de evaluatie van een jonge vrouw die wordt verwezen voor hyperandrogene symptomen.
in een studie van Arlt et al., patiënten met NCCAH werden gekenmerkt door significant hogere BMI en lagere uiteindelijke lengte in vergelijking met overeenkomende controles (41). Deze gegevens, in combinatie met een paar studies die wijzen op de aanwezigheid van insuline ongevoeligheid, suggereren een ongunstig metabolisch profiel dat pathofysiologisch kan worden verklaard door het effect van verhoogde androgeenconcentraties en/of als gevolg van glucocorticoid therapie (42, 43). Osteoporose kan ook bij deze proefpersonen worden gedetecteerd, waarschijnlijk als gevolg van de corticosteroïdentherapie (34).
NCCA-behandeling vanaf de eerste manifestatie tot het volwassen leven
zodra de diagnose van NCCA is vastgesteld, verdienen verschillende kwesties in verband met latere behandelingsaanbevelingen overweging. De eerste vraag die moet worden beantwoord is of behandeling met glucocorticoïden (GC) geïndiceerd is. De Algemene redenering achter deze benadering is dat door voldoende cortisolconcentraties aan de patiënt te verstrekken, haar dagelijkse behoeften worden gedekt en, bijgevolg, CRH-ACTH-asstimulatie zal worden verminderd, leidend tot verminderde bijnierandrogenproductie. De algemene regel met betrekking tot deze benadering is dat GCs bij vervangingstherapie worden gegeven en niet bij farmacologische doses, terwijl de invloed van leeftijd, geslacht, laboratoriumgegevens, patiëntspecifieke aanbevelingen en behandelingsdoelen op glucocorticoïdvervangingstherapie serieus in overweging worden genomen. We moeten ons er echter ook van bewust zijn dat langdurige GC-substitutietherapie kan leiden tot hypercortisolemie, met de resulterende goed gedocumenteerde nadelige effecten op elk aspect van het metabolisme, met name de groei, vetverdeling en insulineresistentie, evenals op psychologisch profiel. Een ander groot nadeel van deze benadering is het ontbreken van een adequate klinische index of biochemische marker van adequate vervangende dosering, zoals er bestaat met betrekking tot TSH-waarden bij hypothyreoïdie.
ten slotte bemoeilijken het feit dat er geen consensus is over het gebruik van GC en het ontbreken van langetermijngegevens over de verschillende modaliteiten van GC-toediening de beslissing van behandelende artsen over en de keuze van de optimale keuze voor elke patiënt. Hydrocortison wordt typisch gebruikt in kinderen, aangezien het het dichtst op het natuurlijke hormoon (cortisol) lijkt, maar het wordt niet beschouwd als een geschikte benadering in adolescenten en jonge wijfjes toe te schrijven aan de behoefte aan veelvoudige dagelijkse het doseren. Vandaar, geven de meeste volwassen endocrinologen de voorkeur aan dexamethason of prednisolon, bij aangewezen dosissen. Anderzijds moet erop worden gewezen dat de gelijkwaardigheid van verschillende GCs gebaseerd is op hun ontstekingsremmende werking en niet op verschillende aspecten van het menselijk metabolisme. Aangezien cortisol bijna 20% van het menselijke genoom beà nvloedt, worden de diverse reacties van verschillende GCs verwacht in diverse weefsels. Deze perceptie wordt bevestigd door het feit dat de toediening van dexamethason aan patiënten met CAH een verslechtering van de indices van insulineresistentie heeft laten zien in vergelijking met andere GCs (44). Bovendien heeft de toediening van 2,5-7,5 mg prednisolon, een als normaal beschouwde dosis, een langdurige negatieve invloed op het botmetabolisme (45). Daarom moet hydrocortison beschouwd worden als de beste vorm van behandeling in geval van GC-suppletietherapie. De komst van de nieuw gesynthetiseerde hydrocortison formule met één pil per dag en de eerste positieve resultaten bij patiënten met CAH toont veel belofte voor de toekomst (46).
behandeling tijdens de kindertijd
NCCAH patiënten die tijdens de kindertijd zijn gediagnosticeerd met tekenen van PP kunnen worden behandeld met hydrocortison met als doel het onderdrukken van de bijnierhormonen en het voorkomen van snelle vooruitgang van de botleeftijd die de uiteindelijke lengte kan beïnvloeden. Bij de patiënten bij wie 1 jaar voor het begin van de puberteit met hydrocortison werd begonnen en die een botleeftijd van minder dan 9 jaar hadden, bleef de uiteindelijke lengte binnen het genetische potentieel (47). Beschikbare studies tonen aan dat de lengte van de volwassen patiënt de verwachte streeflengte benaderde bij patiënten die nauwgezet werden gecontroleerd en die zich strikt hielden aan de medicatieplannen (48-50). Een recente kleine studie van vijf gevallen (patiënten van 6,1–9,2 jaar) toonde aan dat een ultralage dosis dexamethason een veelbelovende optie is om endogene stress en de effecten ervan te verminderen. Of dit een realistische benadering is en hoeveel de toegediende dosis moet worden opgehelderd, moet nog worden opgehelderd (51). Een studie van Nebesio et al. vergeleken de hormonale effecten en farmacokinetiek van hydrocortison, prednisolon en dexamethason bij 9 prepuberale patiënten met CCAH. Zij toonden aan dat dexamethasone machtiger was dan de andere vormen in het bereiken beduidend lagere bijnierhormoonniveaus, vandaar die suggereren dat dexamethasone voor de afschaffing van bijnierandrogen productie effectiever is (46). Natuurlijk zijn verdere studies nodig om deze bevinding te verifiëren of te verwerpen. Voorts, in sommige gevallen, kunnen de opgeheven androgen concentraties tot secundaire stimulatie van de GnRH-as leiden, die tot premature puberteit leiden. In een dergelijk geval kan een parallelle loop met GnRH-analoog worden voorgeschreven als de botleeftijd aanzienlijk hoger is dan de chronologische leeftijd en/of de geprojecteerde eindlengte niet in verhouding staat tot de doelhoogte.
een andere belangrijke overweging betreft gevallen van gelijktijdige GH-deficiëntie in het geval dat behandeling met GnRH-analogen en GH-voorschrift resulteerde in het bereiken van de streeflengte (52). Volgens de huidige richtlijnen wordt de hierboven beschreven therapie met voorzichtigheid alleen aanbevolen voor die gevallen waarin de voorspelde lengte SD -2,25 < de doelhoogte of zelfs lager is (53). Een alternatieve indicatie voor het starten van hydrocortisonbehandeling is een ontoereikende cortisolrespons na ACTH-stimulatie (36).
de voorkeursbehandeling voor GC bij kinderen is gewoonlijk hydrocortison 10-15 mg / m2, verdeeld over drie doses. Vaak zijn lagere doses ook effectief gebleken, te beginnen bij 6 mg / m2 / dag (34). Overdosering moet worden vermeden, gezien het dat kan leiden tot een slechte groei en Cushingoid functies. Regelmatig groeipatroon, een botleeftijd die compatibel is met chronologische leeftijd, en afwezigheid van centrale obesitas kunnen ook dienen als klinische indices voor een passend beheer. Met betrekking tot biochemisch/ hormonaal profiel worden androstenedione en testosteronniveaus in het midden tot bovenste bereik voor botleeftijd beschouwd als betere markers van adequate GC-vervangingstherapie bij kinderen met NCCA. Onderdrukte testosteronspiegels werden echter gevonden bij 10% van de NCCAH-patiënten, terwijl nog eens 28% van de patiënten verhoogde testosteronconcentraties had, wat mogelijk toe te schrijven is aan hydrocortisonvariabiliteit (54, 55). DHEAS waarden worden verlaagd met zeer kleine doses, terwijl de onderdrukking van 17 OHP en progesteron niveaus vereist zeer hoge doses GC. Opgemerkt moet worden dat 17 OHP waarden cruciaal zijn voor de diagnose, maar niet nuttig zijn tijdens de follow–up. Van nota, bloedbemonstering voor hormonale evaluatie moet worden uitgevoerd zonder stopzetting van de therapie. Echter, vanwege de verwarring van de ziekte en de veelzijdige aard ervan, zijn er geen specifieke richtlijnen voor het tijdstip van verandering van het regime of het staken van de behandeling met glucocorticoïden bij kinderen.
behandeling tijdens de adolescentie
patiënten die sinds de kindertijd worden behandeld
totdat het normale menstruatiepatroon bij NCCA-meisjes is vastgesteld, wordt het voortzetten van GC ‘ s die tijdens de kindertijd zijn begonnen sterk aanbevolen (56). Adolescente patiënten vertonen echter vaak onvoldoende naleving van chronische toediening van geneesmiddelen en laten vaak doses achterwege. Bovendien daalt tijdens de puberteit de halfwaardetijd van hydrocortison met 50% als gevolg van verhoogde IGF-1-niveaus, die 11ßOHSD-activiteit vermindert, evenals als gevolg van verhoogde cortisolklaring als gevolg van versterking van glomerulaire filtratiesnelheid (57). Bij de jonge vrouw, 2 jaar na de menarche en als normale ovulatoire cycli zijn geregistreerd, wordt een patiëntgerichte benadering van de hyperandrogene symptomen die kunnen verschijnen sterk aanbevolen.
als er ernstige hyperandrogene bevindingen zijn, zoals hirsutisme, acne en/of oligomenorroe, zal voortzetting van de behandeling worden overwogen. Het is belangrijk om de kwetsbare gevoeligheden van een jonge adolescent te onthouden, die ernstig zal worden beschadigd door de “afstotende” tekenen van hyperandrogenisme. De combinatie van hyperandrogene symptomen, menstruatiestoornissen en slechte kwaliteit van leven is goed gedocumenteerd en is vooral hoog bij jongere vrouwen. Een ander punt dat moet worden overwogen is de reactie van bijnierreserve post ACTH stimulatie. Aangezien de cortisolpiek van puberale en volwassen vrouwen na stimulatie lager is dan 496 nmol / L, moet in geval van stress een behandeling met steroïden worden toegediend (58). Aan de andere kant, als geen van de bovengenoemde symptomen bij de jonge patiënt wordt aangetroffen, kan de GC-behandeling worden gestaakt, waarna regelmatige follow-up wordt geadviseerd.
vrouwen met de eerste manifestatie tijdens de adolescentie of Hyperandrogene symptomen na stopzetting van de behandeling
in de aanwezigheid van hyperandrogene symptomen wordt een patiëntgerichte benadering ten zeerste aanbevolen, gericht op de belangrijkste klachten van de patiënt. Dit is veel beter dan een algemene universele modus operandi, aangezien bij deze laatste benadering de jonge patiënt teleurgesteld zal zijn, wat leidt tot stopzetting van de behandeling en als gevolg daarvan tot nadelige gezondheidsuitkomsten op volwassen leeftijd. In gevallen met ernstig hirsutisme is een kuur van 6 tot 12 maanden met orale anticonceptiepillen (OCP ‘s) een redelijke procedure gezien de gunstige effecten van OCP’ s op de leverproductie van SHBG en de afname van androgeen-afgifte uit de eierstok. Klinische verbetering zal worden verwacht na ten minste 2-3 maanden van OCP ‘ s initiatie, terwijl het gelijktijdige gebruik van een progestageen met minimale androgene eigenschappen sterk wordt aanbevolen. Als OCP ‘ s falen als de eerstelijnsbenadering, kunnen antiandrogenen (spironolacton, flutamide, bicalutamide, cyproteronacetaat en finasteride) aan de behandeling worden toegevoegd. In onze ervaring heeft de toediening van bicalutamide een significante verbetering bereikt in gevallen van ernstige acne, maar vergelijkbare resultaten werden niet verkregen in gevallen met ernstig hirsutisme. Cosmetische benaderingen zoals lasertoepassing en ontharingsmiddelen kunnen ook voor vrouwen worden voorgesteld die van bovenmatige of ongewenste haargroei of hoofdhaarverlies klagen (androgene alopecia) (32, 34, 57).
bij patiënten met onvoldoende cortisolsecretie na stimulatie, of als OCP ‘ s en antiandrogenen niet kunnen worden verdragen, wordt een kuur met GCs ten zeerste aanbevolen (57). Gegevens van New et al. geef aan dat de onregelmatige menses en de acne binnen 3 maanden na de initiatie van de glucocorticoid therapie worden omgekeerd, terwijl het hirsutisme bijna 30 maanden (59) vereist. In tegenstelling tot de kindertijd worden in de adolescentie vaak langerwerkende steroïden gebruikt en worden regimes van 5 mg prednisolon of 0,25 mg dexamethason aanbevolen (53). Echter, in de echte wereld klinische gegevens hebben aangetoond dat een verscheidenheid van verschillende regimes toegepast in NCCAH management (41).
het primaire doel van de behandeling moet zijn dat de patiënt verlichting krijgt van de hyperandrogene symptomen. Dus, bij personen met NCCAH gediagnosticeerd uitsluitend op basis van laboratoriumresultaten, klinische kenmerken moeten begeleiden management aanbevelingen, omdat hormonale en moleculaire bevindingen niet noodzakelijk klinische ernst te voorspellen. Volgens de richtlijnen van de endocriene samenleving moeten NCCAH-patiënten de mogelijkheid krijgen om GC-therapie te staken wanneer de symptomen verdwijnen (60). Natuurlijk mogen deze patiënten niet verloren gaan voor de follow-up, terwijl de behandeling opnieuw moet worden gestart in het geval van herhaling van de symptomen. Verder moeten patiënten in geval van stopzetting worden geïnformeerd over de mogelijkheid van onvruchtbaarheid en moeten zij worden aangemoedigd om medisch advies in te winnen als zij zwanger willen worden (19). Let op: de juiste overdracht van de patiënt van de pediatrische naar de volwassen endocrinoloog moet worden uitgevoerd, optimaal na 1 jaar gesynchroniseerde monitoring (53, 61).
NCCA en reproductie
subfertiliteit
subfertiliteit wordt vaak gerapporteerd in klassieke vormen van CAH, wat voornamelijk te wijten is aan menstruatiestoornissen, chronische anovulatie en anatomische misvormingen. Het geboortecijfer wordt geschat op 17% in vergelijking met 65% van de controlepopulatie (62). Ondertussen, gegevens met betrekking tot de vruchtbaarheid bij vrouwen met NCCAH zijn onlangs in detail beoordeeld en de geschatte onvruchtbaarheid incidentie is 11% Onder NCCAH vrouwen, dat wil zeggen, relatief milder dan in CAH, en in veel gevallen is gemakkelijk op te lossen. Bidet et al. geëvalueerd vruchtbaarheid bij 190 vrouwen die lijden aan NCCAH, 95 van wie wilde zwanger te worden. In deze populatie werden 187 zwangerschappen bij 85 vrouwen gemeld, wat resulteerde in 141 geboortes bij 82 individuen. Er moet worden benadrukt dat 99 van de zwangerschappen (52,9%) plaatsvond vóór de diagnose van NCCAH, drie van hen met de toepassing van ovulatie inductieprotocollen, de rest spontaan, terwijl 88 zwangerschappen vond plaats na nccah diagnose. In de overgrote meerderheid van hen ontwikkelde de zwangerschap zich na de instelling van therapie met hydrocortison, terwijl het bij 11 vrouwen spontaan gebeurde (63, 64). In een ander rapport van 22 waargenomen NCCAH-vrouwen die zwanger wilden worden, volgden 12 zwangerschappen met prednison (65).
miskramen
In het cohort van Bidet et al. het percentage miskramen was 6,5 en 26,3% bij patiënten behandeld met respectievelijk GCs en onbehandelde patiënten (64). De uitkomst van de zwangerschap kan zelfs succesvol zijn zonder glucocorticoïde behandeling in gevallen waarin NCCAH nog niet werd gediagnosticeerd, zoals ook gemeld in een case report door Cuhaci et al. Niettemin ervoer hetzelfde paar twee miskramen en meldde subfertiliteit na deze eerste zwangerschap (66). Wat de mogelijkheid van premature zwangerschappen betreft, zoals beoordeeld door Bidet et al., het verschilt niet significant van die van de rest van de bevolking (15).
Fertiliteitsplanning
voor vrouwen met symptomatisch hyperandrogenisme of met gemelde onvruchtbaarheid, maar die zwanger willen worden, wordt GC-therapie sterk aanbevolen. Gedurende deze periode zijn de gebruikte glucocorticoïden prednison of hydrocortison. De belangrijkste actie, omdat het de volgende stappen zal begeleiden, is genetische tests van de toekomstige ouders. Vrouwen met NCCAH die verlangen om zwanger te worden moeten zich bewust zijn van het risico van het baren van een kind met de klassieke vorm van de ziekte. De uitkomst van zwangerschappen bij vrouwen met NCCAH, en meer specifiek de incidentie van zuigelingen geboren met CCAH, wordt geschat in recente studies tussen 1,5 en 2,5%, met NCCA op ongeveer 15% (36, 64). Deze frequentie hangt natuurlijk af van de referentiepopulatie, een hogere incidentie die voorkomt bij populaties met een hoge mate van interhuwelijken (36). De voorspelde kans dat ouders bevallen van een kind met CAH is 1 op 120 voor onbekend ouderlijk genotype, nul als de vader geen relevante mutatie heeft, en 1 op 4 als hij een heterozygote mutatie heeft (19). Daarom is genetische tests van de partners van deze vrouwen essentieel om het risico van de bevalling van een kind met de klassieke vorm van CAH te beoordelen (64, 67).
in het geval dat de toekomstige moeder twee NC-mutaties draagt (Tabel 1), is er geen absolute noodzaak voor genetische tests bij de toekomstige vader, aangezien, als hij heterozygoot is voor de CYP12A2-mutaties, de nakomelingen het risico lopen om in de toekomst de niet-klassieke vorm van de ziekte te ontwikkelen. Er moet echter worden opgemerkt dat de dragerstatus 2-15% is in verschillende populaties en de helft van deze individuen draagt een ernstige mutatie. Bovendien zijn definitieve aanbevelingen met betrekking tot situaties waarin genetische tests niet vereist zijn, moeilijk gezien de onvolmaakte genotype-fenotype correlatie, met name voor mildere mutaties . De p30l-mutatie, die het vaakst geassocieerd wordt met NCCAH, komt bijvoorbeeld ook voor bij patiënten met klassieke CAH . Een grote multicenter Europese studie toonde onlangs aan dat dit het geval was bij 78% van de patiënten . In deze studie werd de milde v218l-mutatie in 30% van de gevallen geassocieerd met klassiek in plaats van NCCA. Daarom moeten alle patiënten met NCCAH genetische counseling en moleculaire beoordeling van reproductieve partners worden aangeboden.
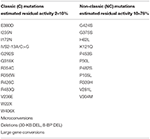
Tabel 1. Mutaties van CYP21A2 met minimale (C) of matige (NC) residuele enzymactiviteit (14, 68, 69).
daarentegen, in het geval dat de toekomstige moeder een samengestelde heterozygoot is met één ernstige (C) en één NC-mutatie, is het genetisch testen van de toekomstige vader verplicht. We moeten in gedachten houden dat gegevens afkomstig van studies met behulp van immunoassays of de meer nauwkeurige LC-MS/MS procedures, consequent wijzen op het onvermogen om betrouwbaar uit te sluiten heterozygositeit met behulp van basale of ACTH gestimuleerd 17 OHP waarden, als gevolg van de significante overlapping met het normale bereik. Men kan het gebruik van een Synacthen-test suggereren, en als deze niet compatibel is met heterozygositeit (som van basale en piekgestimuleerde 17 OHP-waarden < 1,5 nmol/L), dan kan DNA-test worden vermeden. We moeten echter zeer voorzichtig zijn bij de interpretatie van deze test, aangezien deze in andere studies niet is gevalideerd. Vandaar, kan het gebruik van ACTH bevorderde 21-deoxycortisol of afzonderlijk of als deel van een steroid verhouding of steroid profiel, de biochemische identificatie van heterozygotes in de toekomst vergemakkelijken, in het bijzonder aangezien lc-lidstaten/lidstaten wijdverbreid beschikbaar wordt . Als de toekomstige vader een NC mutatie draagt, dan is niets anders nodig. Omgekeerd, als hij drager is van een klassieke mutatie, zal de vraag betreffende de prenatale behandeling van de foetus rijzen. Alle mogelijke genotype combinaties en de voorgestelde procedures worden weergegeven in Tabel 2.

Tabel 2. Planning van voorgestelde procedures tijdens de zwangerschap op basis van het genotype van toekomstige ouders.Prenatale behandeling van CAH Prenatale behandeling van CAH
Het doel van prenatale behandeling van CAH is het voorkomen van genitale virilisatie van de foetus, maar ook het verlichten van de stress die wordt gevoeld door de ouders die waarschijnlijk een kind met dubbelzinnige genitaliën zullen krijgen (70). Dexamethason wordt gebruikt vanwege zijn vermogen om de placenta te passeren en omdat het niet wordt gedeactiveerd vanuit de placenta 11β OH steroïde dehydrogenase en zich slechts minimaal bindt aan het cortisolbindend globuline (CBG) in het bloed van de moeder. Dexamethason, door foetale ACTH secretie te onderdrukken, verlaagt verhoogde foetale androgeenproductie. De behandeling moet worden stopgezet in het geval dat karyotype-of DNA-analyse respectievelijk een mannetje of een onaangetast vrouwtje aan het licht brengt (71). Niettemin, hoewel foetale genitale virilization reeds bij 6-7 weken na conceptie is begonnen, kunnen chorionic villous biopten voor genetische diagnose niet vóór de 10e−12e week worden verkregen. Dit tijdsinterval suggereert dat alle zwangerschappen met een risico op virilizing CAH moeten worden behandeld, ook al is slechts 1 op de 8 foetussen aangetast en vrouwelijk (19, 34, 70).
of deze blootstelling van de foetus aan dexamethason voor preventieve maatregelen medisch en ethisch aanvaardbaar is, blijft controversieel (70). Dierstudies hebben aangetoond dat het eerste trimester dexamethason het geboortegewicht vermindert, de bètacelfunctie van de nier pancreas en de hersenontwikkeling beïnvloedt, en predisponeert voor angst, hypertensie en hyperglycemie op volwassen leeftijd (70). Bovendien klagen 10-20% van zwangere vrouwen die dexamethasonbehandeling gebruiken tijdens de zwangerschap over gewichtstoename, verhoogde eetlust, stemmingswisselingen, slapeloosheid, oedeem, hypertensie, hyperglycemie en striae. Echter, New et al. de Prader-score was lager dan gemiddeld bij foetussen die prenataal met dexamethason werden behandeld, maar er was geen verschil in langetermijnuitkomsten (72). Bovendien, in een beoordeling door Merce Fernandez-Balsells et al. er werd aangetoond dat dexamethason geassocieerd is met een vermindering van de virilisatie van de foetus zonder significante maternale of foetale bijwerkingen (63). De auteurs van het bovengenoemde overzicht en de opeenvolgende richtlijnen van de endocriene samenleving wijzen er echter op dat vanwege de kleine steekproefomvang in de gehele literatuur het onderwerp onzeker blijft en verder onderzoek duidelijk nodig is.
de beslissing over het initiëren van de behandeling dient alleen te worden genomen in grote centra met een ervaren team en protocollen die zijn goedgekeurd door institutionele Beoordelingscommissies en gebaseerd zijn op de waarden en voorkeuren van de familie en met hun schriftelijke geïnformeerde toestemming als voorwaarde (19, 63). Verschillende specialisten op dit gebied geven aan dat een hogere waarde moet worden toegekend aan het voorkomen van onnodige prenatale blootstelling van moeder en foetus aan Dexamethason in plaats van het opleggen van de emotionele tol van dubbelzinnige genitaliën aan ouders en patiënten. Het belangrijkste is dat ouders moet worden verduidelijkt dat toediening van dexamethason de patiëntstatus niet wijzigt, maar gericht is op het verminderen van de noodzaak van een operatie in plaats van het leven of de intellectuele capaciteit in stand te houden. Ze moeten ook weten dat de kans dat hun kind lijdt aan de klassieke vorm van de ziekte hoog is, ondanks de behandeling. Het meest cruciale is dat de blootstelling van een jong organisme aan een zeer krachtig GC tijdens een bijzonder gevoelige periode van foetale programmering en groei, die heel goed nutteloos zou kunnen blijken in het geval van een foetus met het XY karyotype, op dit moment niet wordt ondersteund door robuuste en onbetwistbare gegevens.
idealiter zou er een vroege diagnose moeten zijn, deze wordt uitgevoerd via een niet-invasieve techniek en vóór het begin van de genitale virilisatie van de foetus. In deze richting wijzen nieuwe studies op het gebruik van celvrij foetaal DNA verkregen uit maternaal plasma als een veelbelovende methode die de bepaling van het foetale geslacht en de diagnose van CAH bij een vroege zwangerschapsduur mogelijk maakt (<9 weken). Indien deze procedure op grote schaal wordt toegepast in de klinische praktijk, zal onnodige prenatale behandeling met Dexamethason worden vermeden (73).
behandeling tijdens de zwangerschap
tijdens de zwangerschap dienen vrouwen met NCCAH bij voorkeur met hydrocortison te worden behandeld, niet alleen omdat zwangerschap doorgaans gepaard gaat met verhoogde stressniveaus, maar ook als preventieve maatregel tegen de bovengenoemde hoge incidentie van miskramen. Deze vorm van glucocorticoid wordt begunstigd wegens zijn metabolisatie door placentaenzym 11β OH steroid dehydrogenase II, die glucocorticoid verhindert enig effect op de foetus te hebben (71). Op het moment van de conceptie moet de hydrocortison-dosis worden verhoogd tot 20-25 mg/dag en dosisaanpassingen worden elke 6-8 weken uitgevoerd om de testosteronspiegels op de bovenste normale niveaus van het zwangerschapstrimester te houden (74). Als voorbeeld worden de testosteron-en D4-waarden van een patiënt met NCCAH weergegeven vanaf de conceptie tot 3 jaar na de bevalling van twee gezonde tweelingen (figuur 1).
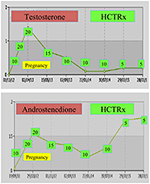
figuur 1. Testosteron-en andostenedionspiegels en daaropvolgende hydrocortisondosering (HCTRx), vanaf de conceptie tot 3 jaar na de bevalling toegediend aan een patiënt met NCCAH.
andere speciale problemen
stressmanagement bij NCCAH
tijdens ernstige levensbedreigende stress, chirurgie of ernstige ziekte kunnen patiënten met NCCA die met glucocorticoïden worden behandeld Grotere of frequentere doses glucocorticoïden nodig hebben, aangezien hun bijnierfunctie iatrogenisch wordt onderdrukt. Daarom is het van cruciaal belang dat ouders van jonge kinderen worden voorgelicht en dat patiënten opnieuw worden voorgelicht over de overgang naar volwassenenzorg. Voor NCCAH-patiënten die niet worden behandeld met GCs of in het geval van stopzetting, moet de cortisolrespons op ACTH worden beoordeeld. Bijna een derde van de NCCAH-patiënten reageert onvoldoende op de stimulatie (15, 75). Voor degenen die normaal reageren op ACTH stimulatie wordt geen behandeling met stressdoses aanbevolen (19). Mineralocorticoïdentherapie is in geen van de gevallen vereist, aangezien deze patiënten een normale aldosteronsecretie hebben en geen zoutverspilling ontwikkelen (58). In het geval van hyperthyreoïdie of een toename van de behandeling met levothyroxine wordt de cortisolklaring verhoogd en kan een bijniercrisis optreden (58, 76).
Psychobiologische aspecten in NCCAH
Meyer-Bahlburg et al. hebben in verschillende studies een verminderd psychologisch profiel gemeld bij patiënten met NCCAH als gevolg van 21oh deficiëntie. Meer specifiek hadden de getroffen vrouwen een hogere masculinization / defeminization score op verschillende maten van geslacht-gerelateerd gedrag in vergelijking met normale controle vrouwen, hoewel duidelijk minder dan bij vrouwen met klassieke CAH. Ze hadden ook een aanzienlijk verhoogde biseksuele of homoseksuele geaardheid. Retrospectieve klinisch-kwalitatieve interviews met deze vrouwen onthulde een geschiedenis van ongemak en sociale stress met betrekking tot hun voorbehandeling ervaringen met androgeen-afhankelijke tekenen, zoals acne, hirsutisme, en conceptie moeilijkheden (59, 77, 78). Ook Arlt et al. toonde aan dat de subjectieve gezondheidstoestand in alle acht domeinen van een kortdurend gezondheidsonderzoek significant was aangetast, met de meest opvallende verschillen in vergelijking met leeftijdsgebonden en geslachtelijke controles, met betrekking tot de domeinen algemene gezondheid, vitaliteit en rolbeperkingen als gevolg van emotionele problemen. De vragenlijst van de Hospital Anxiety and Depression Scale (HADS) toonde ook verhoogde angstscores aan (41). Met het oog op deze bevindingen moeten psychologische parameters als leidraad voor de therapie worden overwogen bij vrouwen met NCCAH en, in de context van de patiëntgerichte aanpak, moet een psychologische diagnose en ondersteuning worden aangeboden.
conclusies
NCCA wordt beschouwd als de frequentere en mildere vorm van CAH vanwege de retentie van 20-50% enzymactiviteit. De meeste patiënten kunnen medisch advies in elk stadium van hun leven als gevolg van symptomen met betrekking tot androgeen overmaat. Deze omvatten voortijdige puber, snelle groei, hirsutisme, acne, menstruele onregelmatigheden, of onvruchtbaarheid, evenals vele andere minder prominente manifestaties. Vroege ochtend baseline waarden van 17 OHP als een goede eerste screening test en verdere evaluatie met ACTH stimulatie en, in het geval van borderline resultaten, genetische testen, wordt aanbevolen. Als algemene regel, androstenedione en niet 17 OHP niveaus moeten worden gebruikt tijdens de follow-up.
De behandelingsbeslissing moet gebaseerd zijn op de beoordeling van de feiten en moet een geïndividualiseerde aanpak volgen. Behandeling is niet altijd geïndiceerd, tenzij de patiënt symptomatisch is, bijvoorbeeld kinderen met een vroeg begin en snelle progressie van schaamhaar en lichaamshaar, snelle groei en/of skeletontwikkeling, of vrouwen met oligomenorroe, acne, hirsutisme, onvruchtbaarheid, of een combinatie van deze en andere van de bovengenoemde symptomen. Genetische counseling wordt sterk aanbevolen bij NCCAH vrouwen die willen zwanger worden, evenals genotypering van de vader. De algehele behandeling van de patiënt omvat bovendien de behandeling van de waarschijnlijke complicaties van glucocorticoïdentherapie of metabolismegerelateerde manifestaties van de ziekte. Ten slotte is het, gezien het feit dat veel therapeutische kwesties met betrekking tot de juiste behandeling van deze patiënten nog niet zijn opgehelderd, van groot belang dat de behandelend arts op de hoogte blijft van alle ontwikkelingen op dit gebied en de nieuwe gegevens in zijn klinische praktijk integreert. Zeker, verdere klinische studies op dit gebied zijn essentieel.
kortom, voor de NCCA-vrouw is de ideale aanpak een op maat gemaakte aanpak, die een soepele overgang van haar management omvat zodra ze wordt doorverwezen van de pediatrische naar de volwassen endocrinoloog, samen met symptoomgerichte behandeling die haar gedurende haar hele leven zal begeleiden. Gezien de veelvoudige factoren die het hyperandrogenic systeem beà nvloeden, is het raadzaam om patiënten aan te moedigen om een gezondere levensstijl aan te nemen door het verbeteren van hun dieetgewoonten, het verhogen van oefening, en het streven naar gewichtsvermindering. Bovendien is psychologische ondersteuning in bepaalde gevallen vaak gunstig. Bovendien hebben specialisten op gebieden die betrokken zijn bij de behandeling van deze aandoening, zoals dermatologen, gynaecologen en psychologen, diepgaande kennis nodig over het beheer van verdachte of reeds gediagnosticeerde gevallen van NCCAH. Laten we tot slot het inzichtelijke citaat van de Amerikaanse astronoom Vera Cooper Rubin in gedachten houden: “de wetenschap vordert het best wanneer waarnemingen ons dwingen onze vooroordelen te veranderen.”
Auteursbijdragen
alle genoemde auteurs hebben een aanzienlijke, directe en intellectuele bijdrage aan het werk geleverd en het voor publicatie goedgekeurd.
belangenconflict verklaring
De auteurs verklaren dat het onderzoek werd uitgevoerd zonder enige commerciële of financiële relatie die als een potentieel belangenconflict kon worden opgevat.
1. Speiser PW, Wit PC. Congenitale bijnierhyperplasie. N Engl J Med. (2003) 349:776–88. doi: 10.1056 / NEJMra021561
PubMed Abstract | CrossRef Full Text/Google Scholar
2. Turcu AF, Auchus RJ. Adrenal steroidogenesis and congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. (2015) 44:275–96. doi: 10.1016/j.ecl.2015.02.002
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Speiser PW. Nonclassic adrenal hyperplasia. Rev Endocr Metab Disord. (2009) 10:77–82. doi: 10.1007/s11154-008-9097-x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Gidlöf S, Falhammar H, Thilén A, von Döbeln U, Ritzén M, Wedell A, et al. Honderd jaar aangeboren bijnierhyperplasie in Zweden: een retrospectieve, populatie-gebaseerde cohort Studie. Lancet Diabetes Endocrinol. (2013) 1:35–42. doi: 10.1016/S2213-8587(13)70007-X
PubMed Abstract | CrossRef Full Text/Google Scholar
5. Speiser PW, Dupont B, Rubinstein P, Piazza a, Kastelan A, New MI. Hoge frequentie van niet-klassieke steroïde 21-hydroxylase deficiëntie. Am J Hum Genet. (1985) 37:650–67.
PubMed Abstract | Google Scholar
6. Wilson RC, Nimkarn S, Dumic M, Obeid J, Azar MR, Azar M, et al. Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Mol Genet Metab. (2007) 90:414–21. doi: 10.1016/j.ymgme.2006.12.005
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Miller WL. Clinical review 54: genetics, diagnosis, and management of 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1994) 78:241–6. doi: 10.1210/jcem.78.2.8106606
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Witchel SF, Azziz R. Nonclassic congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2010) 2010:625105. doi: 10.1186/1687-9856-2010-625105
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Wilson RC, Mercado AB, Cheng KC, New MI. Steroïde 21-hydroxylase deficiëntie: genotype kan het fenotype niet voorspellen. J Clin Endocrinol Metab. (1995) 80:2322–9. doi: 10.1210 / jc.80.8.2322
CrossRef Full Text / Google Scholar
10. Witte PC. Neonatale screening op congenitale bijnierhyperplasie. Nat Rev Endocrinol. (2009) 5:490–8. doi: 10.1038 / nrendo.2009.148
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Kohn B, Levine LS, Pollack MS, Pang S, Lorenzen F, Levy D, et al. Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. (1982) 55:817–27. doi: 10.1210/jcem-55-5-817
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Moran C, Azziz R, Carmina E, Dewailly D, Fruzzetti F, Ibañez L, et al. 21-Hydroxylase deficiënte niet-klassieke bijnierhyperplasie is een progressieve aandoening: een multicenter studie. Am J Verloskundige Gynaecol. (2000) 183:1468–74. doi: 10.1067 / mob.2000.108020
PubMed Abstract | CrossRef Full Text/Google Scholar
13. Azziz R, Hincapie LA, Knochenhauer ES, Dewailly D, Fox L, Boots LR. Screening op 21-hydroxylase deficiënte niet-klassieke bijnierhyperplasie bij hyperandrogene vrouwen: een prospectieve studie. Fertil Steril. (1999) 72:915–25. doi: 10.1016/S0015-0282(99)00383-0
PubMed Abstract | CrossRef Full Text/Google Scholar
14. Livadas S, Dracopoulou M, Dastamani A, Sertedaki A, Maniati-Christidi M, Magiakou A-M, et al. Het spectrum van klinische, hormonale en moleculaire bevindingen bij 280 personen met niet-klassieke congenitale bijnierhyperplasie veroorzaakt door mutaties van het CYP21A2-gen. Clin Endocrinol (Oxf). (2015) 82:543–9. doi: 10.1111 / cen.12543
PubMed Abstract | CrossRef Full Text/Google Scholar
15. Bidet M, Bellanné-Chantelot C, Galand-Portier M-B, Tardy V, Billaud L, Laborde K, et al. Klinische en moleculaire karakterisatie van een cohort van 161 niet-gerelateerde vrouwen met niet-klassieke congenitale bijnierhyperplasie als gevolg van 21-hydroxylase deficiëntie en 330 familieleden. J Clin Endocrinol Metab. (2009) 94:1570–8. doi: 10.1210 / jc.2008-1582
PubMed Abstract | CrossRef Full Text/Google Scholar
16. Escobar-Morreale HF, Sanchón R, San Millán JL. Een prospectieve studie naar de prevalentie van niet-klassieke congenitale bijnierhyperplasie bij vrouwen met hyperandrogene symptomen en tekenen. J Clin Endocrinol Metab. (2008) 93:527–33. doi: 10.1210 / jc.2007-2053
PubMed Abstract | CrossRef Full Text/Google Scholar
17. Dewailly D, Vantyghem-Haudiquet MC, Sainsard C, Buvat J, Cappoen JP, Ardaens K, et al. Klinische en biologische fenotypen bij 21-hydroxylasedeficiëntie met laat begin. J Clin Endocrinol Metab. (1986) 63:418–23. doi: 10.1210 / jcem-63-2-418
PubMed Abstract / CrossRef Full Text / Google Scholar
18. Gönç EN, Ozön ZA, Alikaşifoglu A, Engiz O, Bulum B, Kandemir N. (2011). Is basaal serum 17-OH progesteron een betrouwbare parameter om niet-klassieke congenitale bijnierhyperplasie bij premature adrenarche te voorspellen? Turk J Pediatr. 53:274–80.
PubMed Abstract / Google Scholar
19. Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, et al. Congenitale bijnierhyperplasie als gevolg van steroïde 21-hydroxylase deficiëntie: een endocriene samenleving klinische praktijk richtlijn. J Clin Endocrinol Metab. (2010) 95:4133–60. doi: 10.1210 / jc.2009-2631
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Dacou-Voutetakis C, Dracopoulou M. High incidence of molecular defects of the CYP21 gene in patients with premature adrenarche. J Clin Endocrinol Metab. (1999) 84:1570–4. doi: 10.1210/jc.84.5.1570
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Ambroziak U, Kepczynska-Events I, Kurylowicz, Małunowicz EM, Wójcicka, Miskiewicz P, et al. De diagnose van niet-klassieke congenitale hyperplasie van de bijnierschors als gevolg van 21-hydroxylase deficiëntie, gebaseerd op serum basale of post-ACTH stimulatie 17-hydroxyprogesteron, kan leiden tot vals-positieve diagnose. Clin Endocrinol (Oxf). (2016) 84:23–9. doi: 10.1111 / price 12935
PubMed Abstract | CrossRef Full Text/Google Scholar
22. Ambroziak Y, Kepczynska-Events I, Kurylowicz, Wysłouch-Cieszyńska A, Małunowicz EM, Bartoszewicz, et al. LC-MS/MS verbetert de screening naar 21-hydroxylase deficiëntie. Gynaecol Endocrinol. (2015) 31:296–300. doi: 10.3109 / 09513590.2014.994599
PubMed Abstract | CrossRef Full Text | Google Scholar
23. bbc, CC, Goldman MM, Zhang K, Clark-N, Reitz RE Welt CK. Gelijktijdige meting van dertien steroïde hormonen bij vrouwen met polycysteus ovarium syndroom en controle vrouwen met behulp van vloeibare chromatografie-tandem massaspectrometrie. PLoS ÉÉN. (2014) 9: e93805. doi: 10.1371 / journal.pone.0093805
PubMed Abstract / CrossRef Full Text / Google Scholar
24. Ray JA, Kushnir MM, Yost RA, Rockwood AL, Wayne Meikle A. Performance enhancement in the measurement of 5 endogene steroïden by LC-MS / MS combined with differential ion mobility spectrometry. Clin Chim Acta. (2015) 438:330–6. doi: 10.1016 / j. cca.2014.07.036
PubMed Abstract | CrossRef Full Text/Google Scholar
25. Stoupa a, González-Briceño L, Pinto G, Samara-Boustani D, Thalassinos C, Flechtner I, et al. Onvoldoende cortisol respons op de tetracosactide (Synacthen®) test bij niet-klassieke congenitale bijnierhyperplasie: een uitzondering op de regel? Horm Res Paediatr. (2015) 83:262–7. doi: 10.1159/000369901
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. (1992) 90:584–95. doi: 10.1172/JCI115897
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Temeck JW, Pang SY, Nelson C, New MI. Genetische defecten van steroidogenese in premature puber. J Clin Endocrinol Metab. (1987) 64:609–17. doi: 10.1210 / jcem-64-3-609
PubMed Abstract | CrossRef Full Text/Google Scholar
28. Voutilainen R, Jääskeläinen J. Premature adrenarche: etiology, clinical findings, and consequences. J Steroid Biochem Mol Biol. (2015) 145:226–36. doi: 10.1016 / j.jsbmb.2014.06.004
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Oberfield SE, Sopher AB, Gerken AT. Approach to the girl with early onset of pubic hair. J Clin Endocrinol Metab. (2011) 96:1610–22. doi: 10.1210/jc.2011-0225
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Janus D, Wojcik M, Malunowicz E, Starzyk JB. Een geval van terugkerende labiale adhesie bij een 15 maanden oud kind met asymptomatische niet-klassieke aangeboren bijnierhyperplasie toe te schrijven aan 21-hydroxylase deficiëntie. J Pediatr Endocrinol Metab. (2012) 25:1017–21. doi: 10.1515/jpem-2012-0190
PubMed Abstract | CrossRef Full Text/Google Scholar
31. Gopal-Kothandapani JS, Petkar A, O ‘ Shea E, Banerjee I. Perianal hair as an unusual presentation of non-classical congenital adrenal hyperplasia. BMJ Case Rep. (2013) 2013: bcr2013010123. doi: 10.1136/bcr-2013-010123
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Falhammar H, Nordenström A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine. (2015) 50:32–50. doi: 10.1007/s12020-015-0656-0
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Chrousos GP, Loriaux DL, Mann DL, Cutler GB. Late-onset 21-hydroxylase deficiency mimicking idiopathic hirsutism or polycystic ovarian disease. Ann Intern Med. (1982) 96:143–8. doi: 10.7326/0003-4819-96-2-143
CrossRef Full Text | Google Scholar
34. Witchel SF. Non-classic congenital adrenal hyperplasia. Steroids. (2013) 78:747–50. doi: 10.1016/j.steroids.2013.04.010
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Carmina E, Rosato F, Jannì A, Rizzo M, Longo RA. Extensive clinical experience: relative prevalence of different androgen excess disorders in 950 women referred because of clinical hyperandrogenism. J Clin Endocrinol Metab. (2006) 91:2–6. doi: 10.1210/jc.2005-1457
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Moran C, Azziz R, Weintrob N, Witchel SF, Rohmer V, Dewailly D, et al. Reproductieve uitkomst van vrouwen met 21-hydroxylase deficiënte niet-klassieke bijnierhyperplasie. J Clin Endocrinol Metab. (2006) 91:3451–6. doi: 10.1210 / jc.2006-0062
PubMed Abstract | CrossRef Full Text/Google Scholar
37. Levin JH, Carmina E, Lobo RA. Is de ongepaste gonadotropine secretie van patiënten met polycysteus ovariumsyndroom vergelijkbaar met die van patiënten met aangeboren bijnierhyperplasie bij volwassenen? Fertil Steril. (1991) 56:635–40. doi: 10.1016/S0015-0282(16)54592-0
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Carmina E, Lobo RA. Ovarian suppression reduces clinical and endocrine expression of late-onset congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Fertil Steril. (1994) 62:738–43. doi: 10.1016/S0015-0282(16)56998-2
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Ludwig E. classificatie van de soorten androgenetische alopecia (gemeenschappelijke kaalheid) die in het vrouwelijke geslacht voorkomen. Br J Dermatol. (1977) 97:247–54. doi: 10.1111 / j. 1365-2133.1977.tb15179.x
PubMed Abstract | CrossRef Full Text/Google Scholar
40. Trakakis E, Papadavid E, Dalamaga M, Koumaki D, Stavrianeas N, Rigopoulos D, et al. Prevalentie van niet-klassieke congenitale bijnierhyperplasie als gevolg van 21-hydroxylase deficiëntie bij Griekse vrouwen met acne: een ziekenhuis-gebaseerde cross-sectionele studie. Acad Dermatol Venereol. (2013) 27:1448–51. doi: 10.1111 / j. 1468-3083. 2012. 04613.x
PubMed Abstract / CrossRef Full Text / Google Scholar
41. Arlt W, Willis DS, Wild SH, Krone N, Doherty EJ, Hahner S, et al. Gezondheidsstatus van volwassenen met aangeboren bijnierhyperplasie: een cohortstudie bij 203 patiënten. J Clin Endocrinol Metab. (2010) 95:5110–21. doi: 10.1210 / jc.2010-0917
PubMed Abstract | CrossRef Full Text/Google Scholar
42. Saygili F, Oge A, Yilmaz C. hyperinsulinemie en insuline-ongevoeligheid bij vrouwen met niet-klassieke congenitale bijnierhyperplasie als gevolg van 21-hydroxylase deficiëntie: de relatie tussen serum leptinespiegels en chronische hyperinsulinemie. Horm Res. (2005) 63:270-4. doi: 10.1159/000086363
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Speiser PW, Serrat J, New MI, Gertner JM. Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1992) 75:1421–4. doi: 10.1210/jc.75.6.1421
PubMed Abstract | CrossRef Full Text | Google Scholar
44. Han TS, Stimson RH, Rees DA, Krone N, Willis DS, Conway GS, et al. Glucocorticoïd behandelingsregime en gezondheidsresultaten bij volwassenen met congenitale bijnierhyperplasie. Clin Endocrinol. (2013) 78:197–203. doi: 10.1111 / cen.12045
PubMed Abstract | CrossRef Full Text/Google Scholar
45. Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Use of oral corticosteroids and risk of fractures. J Bone Miner Res. (2000) 15:993-1000. doi: 10.1359 / jbmr.2000.15.6.993
PubMed Abstract | CrossRef Full Text | Google Scholar
46. Nebesio TD, Renbarger JL, Nabhan ZM, Ross SE, Slaven JE, Li L, et al. Differential effects of hydrocortisone, prednisone, and dexamethasone on hormonal and pharmacokinetic profiles: a pilot study in children with congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2016) 2016:17. doi: 10.1186 / s13633-016-0035-5
PubMed Abstract / CrossRef Full Text / Google Scholar
47. Weintrob N, Dickerman Z, Sprecher E, Galatzer a, Pertzelan A. Non-classical 21-hydroxylase deficiency in infancy and childhood: the effect of time of initiation of therapy on puberty and final height. J Endocrinol. (1997) 136:188–95. doi: 10.1530 / eje.0.1360188
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Bonfig W, Bechtold S, Schmidt H, Knorr D, Schwarz HP. Reduced final height outcome in congenital adrenal hyperplasia under prednisone treatment: deceleration of growth velocity during puberty. J Clin Endocrinol Metab. (2007) 92:1635–9. doi: 10.1210/jc.2006-2109
PubMed Abstract | CrossRef Full Text | Google Scholar
49. Dörr HG. Groei bij patiënten met klassieke congenitale bijnierhyperplasie als gevolg van 21-hydroxylase deficiëntie. Horm Res. (2007) 68 (Suppl 5):93-9. doi: 10.1159 / 000110587
PubMed Abstract | CrossRef Full Text/Google Scholar
50. Hoepffner W, Kaufhold A, Willgerodt H, Keller E. patiënten met klassieke congenitale bijnierhyperplasie als gevolg van 21-hydroxylase deficiëntie kunnen hun doelhoogte bereiken: de Leipzig experience. Horm Res. (2008) 70:42-50. doi: 10.1159/000129677
PubMed Abstract | CrossRef Full Text | Google Scholar
51. van der Kaay D, van den Akker E. Ultralow-dose dexamethasone to preserve endogenous cortisol stress response in nonclassical congenital adrenal hyperplasia: a new promising treatment. Int J Endocrinol Metab. (2014) 12:e14657. doi: 10.5812/ijem.14657
CrossRef Full Text | Google Scholar
52. Lin-Su K, Vogiatzi MG, Marshall I, Harbison MD, Macapagal MC, Betensky B, et al. Treatment with growth hormone and luteinizing hormone releasing hormone analog improves final adult height in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. (2005) 90:3318–25. doi: 10.1210/jc.2004-2128
PubMed Abstract | CrossRef Full Text | Google Scholar
53. McCann-Crosby B, Chen M-J, Lyons SK, Lin Y, Axelrad M, Dietrich JE, et al. Nonclassical congenital adrenal hyperplasia: doelen van behandeling en overgang. Pediatr Endocrinol Rev. (2014) 12: 224-38.
PubMed Abstract / Google Scholar
54. Ogilvie CM, Crouch NS, Rumsby G, Creighton SM, Liao L-M, Conway GS. Congenitale bijnierhyperplasie bij volwassenen: een overzicht van medische, chirurgische en psychologische problemen. Clin Endocrinol. (2006) 64:2–11. doi: 10.1111 / j. 1365-2265.2005.02410.x
PubMed Abstract | CrossRef Full Text | Google Scholar
55. Mah PM, Jenkins RC, Rostami-Hodjegan A, Newell-Price J, Doane A, Ibbotson V, et al. Weight-related dosing, timing and monitoring hydrocortisone replacement therapy in patients with adrenal insufficiency. Clin Endocrinol. (2004) 61:367–75. doi: 10.1111/j.1365-2265.2004.02106.x
PubMed Abstract | CrossRef Full Text | Google Scholar
56. Diaz A, Laufer Mr Breech LLAA van PC op A, Care AC van O en GC op AH. Menstruatie bij meisjes en adolescenten: het gebruik van de menstruatiecyclus als een vitaal teken. Kindergeneeskunde. (2006) 118:2245–50. doi: 10.1542 / peds.2006-2481
CrossRef Full Text / Google Scholar
57. Merke DP, Poppas DP. Behandeling van adolescenten met congenitale bijnierhyperplasie. lancet Diabetes Endocrinol. (2013) 1:341–52. doi: 10.1016/S2213-8587(13)70138-4
PubMed Abstract | CrossRef Full Text | Google Scholar
58. Verma S, Green-Golan L, VanRyzin C, Drinkard B, Mehta SP, Weise M, et al. Adrenomedullary function in patients with nonclassic congenital adrenal hyperplasia. Horm Metab Res. (2010) 42:607–12. doi: 10.1055/s-0030-1253385
PubMed Abstract | CrossRef Full Text | Google Scholar
59. New MI. Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2006) 91:4205–14. doi: 10.1210/jc.2006-1645
PubMed Abstract | CrossRef Full Text | Google Scholar
60. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2018) 103:4043–88. doi: 10.1210/jc.2018-01865
PubMed Abstract | CrossRef Full Text | Google Scholar
61. Kruse B, Riepe FG, Krone N, Bosinski HAG, Kloehn S, Partsch CJ, et al. Congenital adrenal hyperplasia – how to improve the transition from adolescence to adult life. Exp Clin Endocrinol Diabetes. (2004) 112:343–55. doi: 10.1055/s-2004-821013
PubMed Abstract | CrossRef Full Text | Google Scholar
62. Gastaud F, Bouvattier C, Duranteau L, Brauner R, Thibaud E, Kutten F, et al. Verminderde seksuele en reproductieve resultaten bij vrouwen met klassieke vormen van congenitale bijnierhyperplasie. J Clin Endocrinol Metab. (2007) 92:1391–6. doi: 10.1210 / jc.2006-1757
PubMed Abstract | CrossRef Full Text/Google Scholar
63. Mercè Fernández-Balsells M, Muthusamy K, Smushkin G, Lampropulos JF, Elamin MB, Abu Elnour NO, et al. Prenatale dexamethasongebruik voor de preventie van virilization in zwangerschappen die risico lopen op klassieke congenitale bijnierhyperplasie wegens 21-hydroxylase (CYP21A2) deficiëntie: een systematische beoordeling en meta-analyses. Clin Endocrinol. (2010) 73:436–44. doi: 10.1111 / j. 1365-2265.2010.03826.x
CrossRef Full Text / Google Scholar
64. Bidet M, Bellanné-Chantelot C, Galand-Portier MB, Golmard JL, Tardy V, Morel Y, et al. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2010) 95:1182–90. doi: 10.1210/jc.2009-1383
PubMed Abstract | CrossRef Full Text | Google Scholar
65. Birnbaum MD, Rose LI. Late onset adrenocortical hydroxylase deficiencies associated with menstrual dysfunction. Obstet Gynecol. (1984) 63:445–51.
PubMed Abstract | Google Scholar
66. Cuhaci N, Aydin C, Yesilyurt A, Pinarli FA, Ersoy R, Cakir B. Nonclassical congenital adrenal hyperplasia and pregnancy. Case Rep Endocrinol. (2015) 2015:296924. doi: 10.1155/2015/296924
PubMed Abstract | CrossRef Full Text | Google Scholar
67. Deneux C, Tardy V, Anne D, Mornet E, Billaud L, Charron D, et al. Correlatie tussen fenotype en genotype bij 56 vrouwen met niet-klassieke congenitale bijnierhyperplasie als gevolg van 21-hydroxylase deficiëntie. J Clin Endocrinol Metab. (2001) 86:207–13. doi: 10.1210 / jcem.86.1.7131
PubMed Abstract | CrossRef Full Text/Google Scholar
68. Simonetti L, Bruque CD, Fernández CS, Benavides-Mori B, Delea M, Kolomenski JE, et al. CYP21A2 mutatie update: uitgebreide analyse van databases en gepubliceerde genetische varianten. Hum Mutat. (2018) 39:5–22. doi: 10.1002 / humu.23351
PubMed Abstract | CrossRef Full Text | Google Scholar
69. OMIM Entry – * 613815 – CYTOCHROME P450 FAMILY 21 SUBFAMILY A POLYPEPTIDE 2; CYP21A2.
Google Scholar
70. Miller WL, Witchel SF. Prenatal treatment of congenital adrenal hyperplasia: risks outweigh benefits. Am J Obstet Gynecol. (2013) 208:354–9. doi: 10.1016/j.ajog.2012.10.885
PubMed Abstract | CrossRef Full Text | Google Scholar
71. Lekarev O, New MI. Adrenal disease in pregnancy. Best Pract Res Clin Endocrinol Metab. (2011) 25:959–73. doi: 10.1016/j.beem.2011.08.004
PubMed Abstract | CrossRef Full Text | Google Scholar
72. New MI, Abraham M, Yuen T, Lekarev O. An update on prenatal diagnosis and treatment of congenital adrenal hyperplasia. Semin Reprod Med. (2012) 30:396–9. doi: 10.1055/s-0032-1324723
PubMed Abstract | CrossRef Full Text | Google Scholar
73. Kazmi D, Bailey J, Yau M, Abu-Amer W, Kumar A, Low M, et al. New developments in prenatal diagnosis of congenital adrenal hyperplasia. J Steroid Biochem Mol Biol. (2017) 165:121–3. doi: 10.1016/j.jsbmb.2016.06.016
PubMed Abstract | CrossRef Full Text | Google Scholar
74. Clayton PE, Miller WL, Oberfield SE, Ritzén EM, Sippell WG, Speiser PW, et al. Consensus statement on 21-hydroxylase deficiency from the European Society for Paediatric Endocrinology and the lawson wilkins pediatric endocrine society. Horm Res. (2002) 58:188-95. doi: 10.1159 / 000065490
PubMed Abstract | CrossRef Full Text | Google Scholar
75. Nandagopal R, Sinaii N, Avila NA, van Ryzin C, Chen W, Finkielstain GP, et al. Fenotypische profilering van ouders met cryptische nonclassische congenitale bijnierhyperplasie: bevindingen in 145 niet-gerelateerde families. J Endocrinol. (2011) 164:977–84. doi: 10.1530/EJE-11-0019
PubMed Abstract | CrossRef Full Text/Google Scholar
76. Takasu N, Nakachi K, Higa H. ontwikkeling van de hyperthyreoïdie van Graves veroorzaakte een bijniercrisis bij een patiënt met niet eerder herkende niet-klassieke 21-hydroxylasedeficiëntie. Stagiair Med. (2010) 49:1395–400. doi: 10.2169 / interne geneeskunde.49.3573
PubMed Abstract | CrossRef Full Text | Google Scholar
77. Meyer-Bahlburg HFL, Dolezal C, Baker SW, Carlson AD, Obeid JS, New MI. Prenatal androgenization affects gender-related behavior but not gender identity in 5-12-year-old girls with congenital adrenal hyperplasia. Arch Sex Behav. (2004) 33:97–104. doi: 10.1023/B:ASEB.0000014324.25718.51
CrossRef Full Text | Google Scholar
78. Meyer-Bahlburg HFL, Dolezal C, Baker SW, Nieuw MI. Seksuele geaardheid bij vrouwen met klassieke of niet-klassieke congenitale bijnierhyperplasie als functie van de mate van prenatale androgeen overmaat. Arch Sex Behav. (2008) 37:85–99. doi: 10.1007 / s10508-007-9265-1
PubMed Abstract / CrossRef Full Text / Google Scholar