a fehérje elsődleges szerkezetét olyan aminosavak szekvenciájaként definiáljuk, amelyekből áll. Ez a szekvencia végső soron meghatározza azt a formát, amelyet a fehérje a fehérje atomjainak elrendezésére vonatkozó térbeli korlátozások, az összetevő aminosavmaradványainak kémiai tulajdonságai, valamint a fehérje környezete alapján fogad el.
az aminosavmaradványokat egy polipeptidben összekapcsoló peptidkötések egy aminosav savas karboxilcsoportja és egy másik aminosav bázikus aminosavcsoportja közötti kondenzációs reakcióban alakulnak ki. A peptid összefüggésében az amidcsoportot (CO–NH) peptidcsoportnak nevezik.
a fehérjeszerkezet megértéséhez elengedhetetlen a peptidkötés szerkezetének ismerete. Linus Pauling az 1930-as években röntgendiffrakciót használt a két aminosav között kialakult peptidkötés természetének vizsgálatára. Beszámolt arról, hogy a peptidcsoport (CO–NH) merev síkszerkezettel rendelkezik. Ez a szerkezet miatt közötti kölcsönhatások elektronok a kettős kötés a karbonil csoport, illetve a C–N bond (2. Ábra) olyan, hogy az utóbbi szerez részleges (40%), kettős-bond tulajdonságok.

Ez a hatás a rezonancia példája, amelyet úgy lehet gondolni, mint az elektronok megosztását a kötések között. Mivel a két atom közötti egyetlen kötés hosszabb, mint az azonos két atom közötti kettős kötés, a Peptidcsoportban a C–N és C=O kötések hossza különbözik azoktól, amelyeket más összefüggésekben figyeltek meg, ahol a rezonancia nem fordul elő. Így a részleges kettős kötés jellege C–N a peptid csoport azt jelenti, hogy ez a kötelék rövidebb lenne megjósolta, hogy a C–N egyetlen kötelék, míg a C=O kötés, hogy egy részleges egyetlen bond karakter miatt rezonancia, hosszabb lenne megjósolni a C=O kettős kötés. A peptidcsoport kötési hosszát a 3. ábra mutatja. Hasonlítsa össze a peptidcsoport C-N kötését az N és Ca közötti kötéssel (az A C atom, amelyhez az aminocsoport és a karboxilcsoport kapcsolódik).

a planáris peptidkötésnek két lehetséges konformációja van: a transz-peptidcsoportban a Ca-atomok a peptidkötés ellentétes oldalán vannak (3a. ábra), a cisz-peptidcsoportban pedig a Ca-atomok a peptidkötés ugyanazon oldalán vannak (3b.ábra).
-
figyelembe véve a cisz-ben lévő atomok térbeli elrendezését és közelségét, valamint a peptidkötés transz-konformációit, mely konformáció szerinted előnyös lenne?
-
a transzkonformáció energetikailag kedvezőbb lenne, mint a cis konformáció, mivel minimalizálja a szterikus akadályt.
általánosságban elmondható, hogy a peptidkötések a transzformációban vannak. A cis formák azonban előfordulhatnak olyan peptidkötésekben, amelyek megelőzik a prolin maradékot. Ilyen esetekben a cis forma a szokásosnál stabilabb, mivel a proline oldallánc kevesebb akadályt kínál. Ennek ellenére a cisz-peptidkötések csak a prolinmaradványokat megelőző peptidkötések körülbelül 10% – ában fordulnak elő.
szem előtt tartva a peptidcsoport sík jellegét, egy polipeptidlánc látható, hogy van egy gerince, amely a Ca atomok által összekapcsolt merev sík peptidcsoportokból áll. A 4. ábra egy polipeptid egy részét mutatja, két planáris peptidcsoporttal a transz-konformációban. Vegye figyelembe, hogy bár a peptidkötések forgása nem megengedett, a Ca–N és Ca–C kötések körül forgási lehetőség van. A forgási szögek, amelyeket torziós szögeknek neveznek, ezekről a kötésekről meghatározzák a polipeptid gerincének konformációját. A Ca-N és Ca–C kötések torziós szögeit ɸ (phi) és ψ néven említik. (psi), illetve 180° – ban vannak meghatározva, amikor a polipeptid a kiterjesztett sík konformációban van, amint azt a 4.ábra szemlélteti.
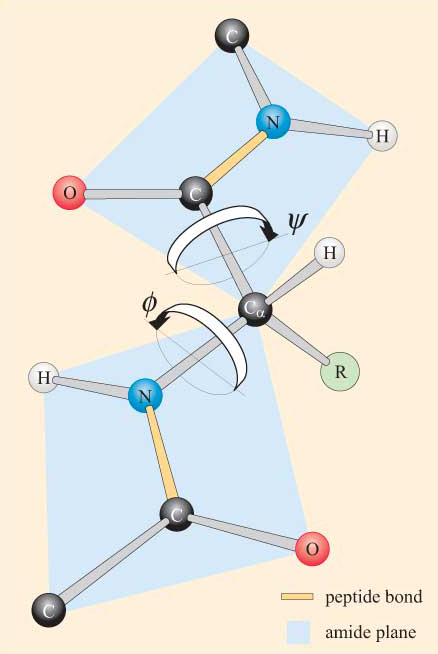
nem lepődsz meg, ha megtudod, hogy a Ster-ra és ψ-ra szterikus korlátok vonatkoznak.
ezeknek a szterikus korlátoknak köszönhetően csak a ɸ És ψ bizonyos értékei, tehát a peptid konformációi megengedettek, míg mások nem.
egy adott szermaradék megengedett értékeit egy polipeptid összefüggésében lehet kiszámítani. Ez a számítás által végzett első meghatározó a távolságok között a nem-kötődés atomok két szomszédos peptid csoportok (például a Ábra 4) a lehetséges értékek ɸ, valamint ψ. Ez a legkönnyebben egy olyan polipeptid esetében történik, amely csak egyfajta aminosavat tartalmaz. A ψ konformációs parcelláját ψ ellen egy adott maradvány számára Ramachandran telekként ismert (feltalálója, G. N. Ramachandran után). Egy ilyen telek lehetővé teszi számunkra, hogy azonosítsuk azokat a conformations (azaz egy adott értéke ɸ, valamint ψ), amelyek sterically kedvező vagy kedvezőtlen (mint az 5. Ábra), a következő kritériumok szerint:
-
Ahol nincs konfliktus a van der Waals sugarak a nem-kötődés atomok, egy alkalmazkodás a ‘szabad’. Ezek a konformációk az 5. ábrán látható kék területeken találhatók.
-
az interatomikus távolságokat a megengedett határértéknél igénylő Konformációk a “külső határ” konformációk. Az 5. ábrán a zöld területeken fekszenek.
-
elméleti konformációk, amelyek megkövetelik, hogy bármely két nem kötő Atom közelebb legyen egymáshoz, mint a van der Waals sugarak lehetővé teszik, szigorúan “tilos”. Ezek az 5. ábrán látható fehér területeken fekszenek.
vegye figyelembe, hogy az 5.ábrán szereplő ɸ És ψ értékek −180º-tól +180º-ig terjednek. A peptidcsoport 360º-on keresztüli elfordítása természetesen visszahozza a kiindulási helyzetbe, és −180º és +180º ugyanannak a helyzetnek felel meg. Így az 5. ábrán a telek bal alsó sarkában lévő zöld csík szomszédos a bal felső sarokban lévő mezővel.

-
használja az 5. ábrát annak meghatározására, hogy a ɸ És ψ következő értékei szterikusan kedvezőek vagy kedvezőtlenek −e: a) ɸ = 90º és ψ = 90º; b) ɸ = – 90º és ψ = 90º.
-
(a) ; B) kedvező.
a Ramachandran parcellák mind a 20 aminosav polimerjeire kialakíthatók. Fontos megjegyezni,hogy sok aminosav-maradék Ramachandran-parcellái általában nagyon hasonlóak, csak három, kedvező vagy tolerálható konformációval rendelkező régió van (az 5. ábrán a poli-L-alanin esetében az 1-3. Különbségek azonban előfordulnak. Például, ha az oldallánc (R A 4. ábrán) a Ca közelében elágazik, mint a treonin esetében, több helyet foglal el a peptid gerincéhez közel, és korlátozza az atomok megközelítését a szomszédos peptidcsoportokban. Ennek eredményeként a megengedett konformációk (ɸ És ψ szögek) korlátozottabbak az elágazó aminosavak polipeptidjeire.
-
a prolin a megengedett konformációk tekintetében is nagyon különbözik a többi aminosavtól, a polifrolin esetében pedig csak a −85º −tól-35º-ig terjedő értékek tolerálhatók. Gondolkodva a proline szerkezetéről, hogyan magyarázhatja meg ezt a viszonylag szűk megengedett ɸ értékeket?
-
a prolin oldallánca kovalensen kötődik az aminocsoport N – hez, így a poliprolinban kevesebb forgási szabadság lesz a Ca–N kötésben, mint más aminosavakkal. Következésképpen az engedélyezett ɸ értékek viszonylag korlátozottak lesznek a többi aminosavhoz képest.
-
6. ábra a polipeptidláncban található glicin-maradványok Ramachandrán-diagramját mutatja. A régiók színkódolása az 5. ábrán látható. Mit mondhatsz a glicin által alkalmazott konformációkról? Tekintsük a glicin szerkezetét. Miért különbözik a glicin a többi maradéktól a konformációk tekintetében?
-
A glicin sokkal nagyobb konformációs szabadsággal rendelkezik, mint más aminosav-maradékok, mert kevésbé erősen akadályozza.

Az egészséges személyeknél telkek számokban 5 6 keletkeztek számára, illetve l-alanin, l-glicin alapján engedélyezett, illetve külső határ távolsága interatomic kapcsolatok meghatározva, az ismert értékek a van der Waals sugarak az atomok (1.Táblázat).
1. táblázat Van der Waals távolságok az interatomikus kapcsolatokhoz.
| általában megengedett/Å | ülső határ/Å | ||
|---|---|---|---|
| H···H | 2.0 | 1.9 |
3.0 |
ezért inkább prediktív, mint tényleges konformációs telkek. Természetesen röntgendiffrakcióval kísérletileg meghatározhatjuk a ɸ És ψ “valódi” értékeit a polipeptidben lévő maradványok esetében. A 7. ábrán az összes maradék ɸ És ψ értékét (a glicin és a prolin kivételével) számos különböző szerkezetben nagy felbontású röntgendiffrakcióval határoztuk meg, és egy Ramachandrán telken ábrázoltuk. Láthatjuk, hogy feltűnő a levelezés az előre jelzett és a tényleges konformációk között. Vegye figyelembe azonban, hogy vannak olyan maradványok, amelyek megfelelnek a “tiltott” területeknek. A maradványok nagy része a 2-es és 3-as “megengedett” régiók közötti régióban található, körülbelül ψ = 0.

-
nézze meg újra a 4. ábrát, és képzelje el, hogy a legfelső peptidcsoportot 180° – on keresztül csavarhatja úgy, hogy ψ = 0. Milyen csoportok valószínűleg ütköznek ebben a konformációban?
-
a szomszédos peptidcsoportok n-H csoportjai ütköznek egymással, szoros közelségbe kényszerítve őket.
az ezekkel a konformációkkal kapcsolatos konfliktusokat a peptidkötés kis mértékű csavarásával lehet megoldani. Így ilyen konformációkban a peptidcsoport ki van csavarva a szokásos planáris konformációból.
bizonyos maradékanyagok korlátozott számú “tiltott” konformációja tolerálható egy polipeptidben, ha az elfogadott konformáció egésze energetikailag kedvező. A polipeptid hajlamos összehajtani, hogy elfogadja a legstabilabb konformációt. Ebben a konformációban a polipeptid minimalizálja szabad energiáját. A következő szakaszokban megvizsgáljuk ezt a magasabb szintű fehérjeszerkezetet.