La esclerótica azul es causada por la delgadez y transparencia de las fibras de colágeno de la esclerótica que permiten la visualización de la úvea subyacente (Figura 1). La esclerótica es la capa externa blanca del ojo, que rodea el iris puede adelgazarse en enfermedades congénitas como la osteogénesis imperfecta 1). Otras enfermedades asociadas con la esclerótica azul incluyen múltiples trastornos del tejido conectivo, como el síndrome de córnea quebradiza 2), el síndrome de Marshall y Stickler 3), el síndrome de POEMS (polineuropatía, organomegalia, endocrinopatía, proteínas monoclonales y cambios en la piel) 4), el síndrome de Marfan, el síndrome de Ehlers–Danlos, el pseudoxantoma elástico y el síndrome de Willems De Vries, por nombrar algunos. Los trastornos óseos y sanguíneos también en la lista incluyen anemia de diamante-Blackfan, anemia por deficiencia severa de hierro, enfermedad juvenil de Paget y deficiencia de fosfatasa ácida 5).
Las formas graves de osteogénesis imperfecta se diagnostican con mayor frecuencia en las primeras etapas de la vida, pero es posible que los casos leves no se noten hasta más adelante en la vida. El color azul grisáceo de la esclerótica se debe a las venas coroideas subyacentes que se ven a través. Esto se debe a que la esclerótica es más delgada de lo normal porque el colágeno defectuoso tipo I no se forma correctamente 6). La esclerótica es una estructura densa de tejido conectivo mal vascularizado compuesta de colágeno de los tipos I, III, IV, V, VI y VIII, así como elastina, proteoglicanos y glicoproteínas 7).
En los Estados Unidos, se estima que la incidencia de osteogénesis imperfecta es de una por cada 20.000 nacidos vivos. Se estima que entre 20,000 y 50,000 personas están afectadas por la osteogénesis imperfecta en los Estados Unidos 8).
Las personas con osteogénesis imperfecta nacen con tejido conectivo defectuoso, o sin la capacidad de producirlo, generalmente debido a una deficiencia de colágeno Tipo I. Esta deficiencia surge de una sustitución de aminoácidos de glicina a aminoácidos más voluminosos en la estructura de triple hélice de colágeno. Como resultado, el cuerpo puede responder hidrolizando la estructura de colágeno inadecuada 9). Si el cuerpo no destruye el colágeno inadecuado, la relación entre las fibrillas de colágeno y los cristales de hidroxiapatita para formar el hueso se altera, causando fragilidad. Como trastorno genético, la osteogénesis imperfecta se ha visto históricamente como un trastorno autosómico dominante del colágeno Tipo I. La mayoría de los casos han sido causados por mutaciones en los genes COL1A1 y COL1A2 10). En los últimos años, se han identificado formas autosómicas recesivas. Se han descrito al menos siete subconjuntos, aunque cuatro subtipos principales son los más comunes y varían de leves a graves. Las personas con osteogénesis imperfecta Tipo I tienen poca deformidad ósea, esclerótica azul persistente, estatura casi normal en la edad adulta y una probabilidad >del 50% de pérdida de audición en la edad adulta. Los pacientes con osteogénesis imperfecta letal perinatal (Tipo II) muestran la mayor gravedad, con fracturas múltiples en el útero o en el parto. Por lo general, estos pacientes nacen muertos o mueren prematuramente. La gravedad de la osteogénesis imperfecta depende del defecto genético específico. La mayoría de los casos de osteogénesis imperfecta se heredan de uno de los padres. Sin embargo, algunos casos son el resultado de nuevas mutaciones genéticas. Una persona con osteogénesis imperfecta tiene un 50% de probabilidades de transmitir el gen y la enfermedad a sus hijos 11).
Figura 1. Anatomía ocular
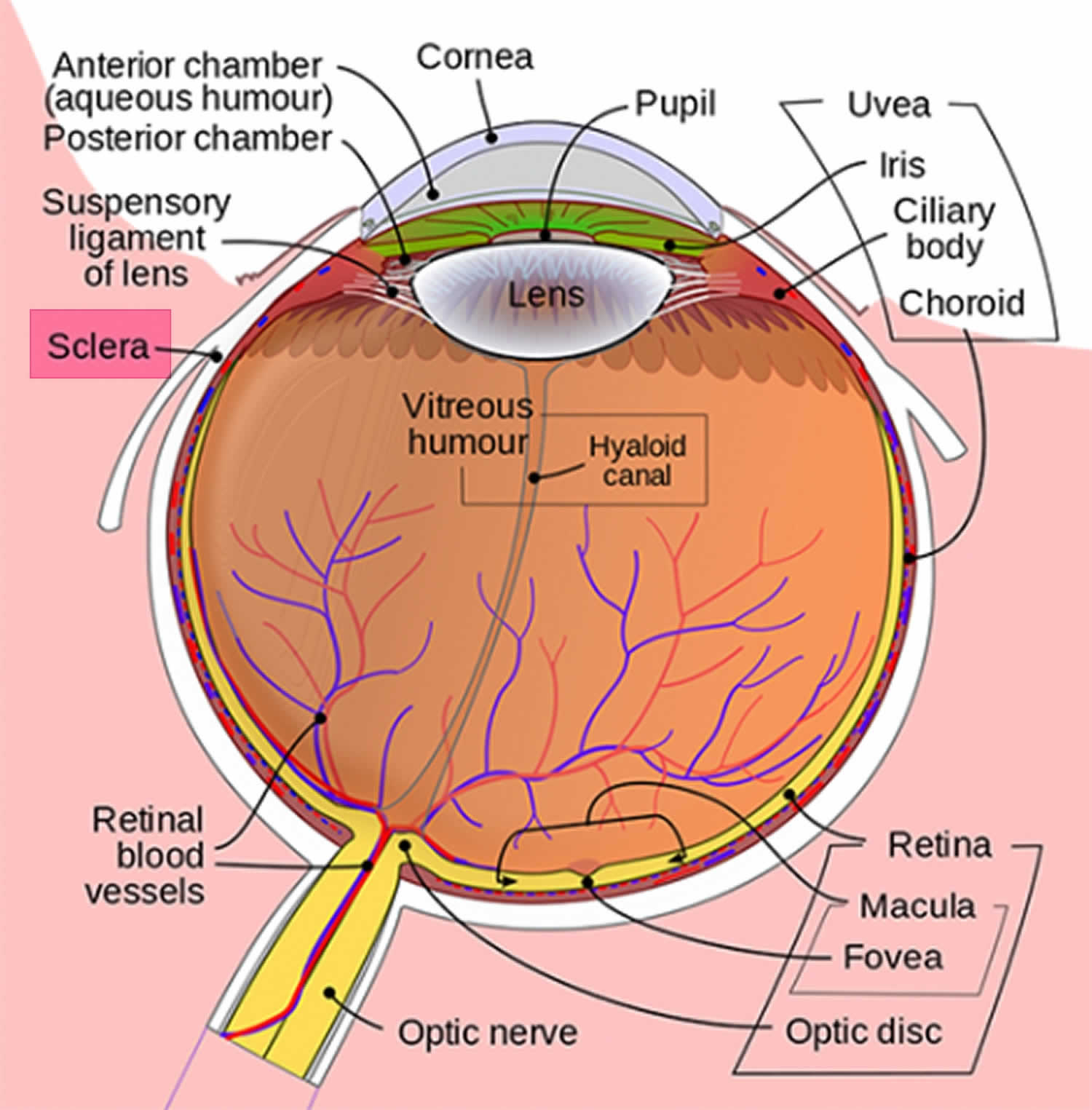
Figura 2. Esclerótica azul
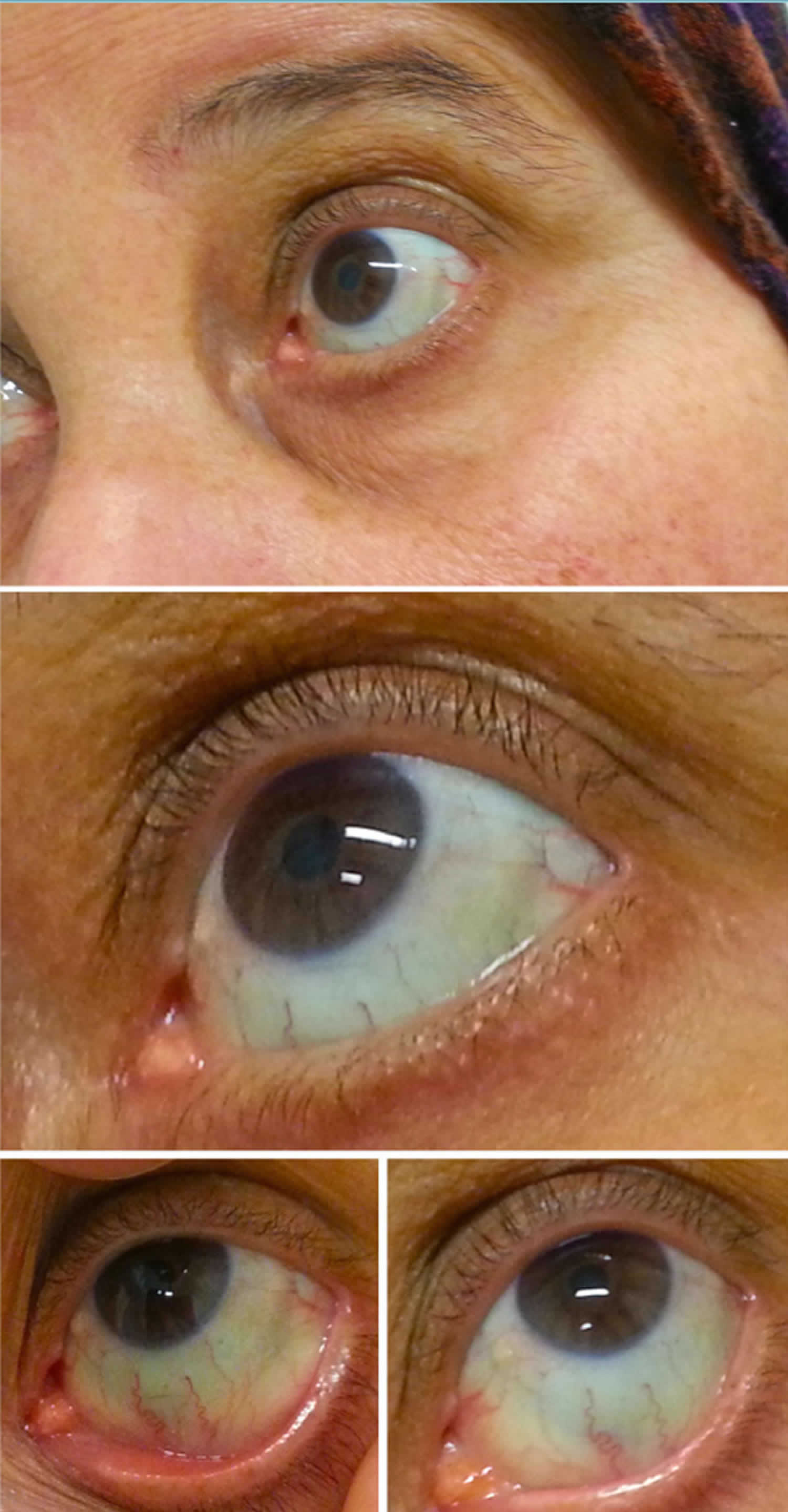
La esclerótica azul causa
La esclerótica azul es la manifestación más consistente de osteogénesis imperfecta, que resulta de una mutación en COLIA1 y COL1A2, que codifica para procolágeno tipo I. Sin embargo, las clasificaciones de esta afección (tipos IV a VI) se han identificado con escleras normales. La osteogénesis imperfecta también se asocia con fragilidad anormal de los huesos y sordera.
La córnea quebradiza, la esclerótica azul y el pelo rojo están asociados con el síndrome de la córnea quebradiza, una afección que también se presenta con anomalías esqueléticas, dentales y cutáneas 13). Se ha encontrado que una mutación sin sentido en ZNF469 es causante de la enfermedad.
Otras afecciones subyacentes asociadas con la formación de esclerótica azul incluyen el síndrome de Ehlers-Danlos (tipo VI), pseudoxantoma elástico, córnea plana, esclerocornea periférica, buftalmos, queratocono, queratoglobus, miopía alta, estafiloma ciliar / ecuatorial, melanocitosis oculodérmica y microcornea. En raras ocasiones, la esclerótica azul se presenta con el síndrome de Hallermann-Streiff, el síndrome de Marfan, el síndrome de Turner, el síndrome de Cheney, el síndrome de Menkes, la picnodisostosis, las córneas quebradizas o la displasia ectodérmica.
La esclerótica azul también puede ocurrir en bebés normales durante los primeros meses de vida; sin embargo, la persistencia de la decoloración azul a lo largo del tiempo puede sugerir la presencia de presión intraocular elevada. Los bebés prematuros con frecuencia presentan escleras azules, particularmente los de origen caucásico.
La esclerótica azul también puede ocurrir de forma aislada como una anomalía hereditaria autosómica dominante o autosómica recesiva 14).
Síntomas de la esclerótica azul
La esclerótica azul se presenta con un aspecto azulado en la esclerótica y puede estar relacionada con una causa patológica o no patológica. Otras características oculares de los trastornos del tejido conectivo asociados con la esclerótica azul incluyen córnea delgada, pliegue epicantal, miopía, queratocono y rayas angioides. Las características sistémicas de los trastornos del tejido conectivo asociados con la esclerótica azul incluyen anomalías de la piel, anomalías cardíacas, cifoescoliosis, hipermovilidad articular, huesos frágiles, anomalías auditivas, anomalías vasculares y anomalías gastrointestinales 15).
Diagnóstico de esclerótica azul
La evaluación diagnóstica de la esclerótica azul incluye un examen externo, biomicroscopia con lámpara de hendidura y evaluación sistémica para trastornos asociados.Aunque no existe una prueba definitiva para la osteogénesis imperfecta, la prueba genética puede confirmar o excluir mutaciones conocidas 16).
Tratamiento de la esclerótica azul
El tratamiento de la esclerótica azul implica el diagnóstico y el tratamiento de la causa subyacente. La esclerótica azul se debe principalmente a síndromes genéticos y, en menor medida, a trastornos no genéticos y puede ocurrir como un efecto secundario de la ingesta de medicamentos. Las escleras azules se asocian comúnmente con trastornos congénitos de la síntesis de colágeno, como la osteogénesis imperfecta, el síndrome de Marfan, el síndrome de Ehlers–Danlos, el pseudoxantoma elástico y el síndrome de Willems De Vries, por nombrar algunos. Los trastornos óseos y sanguíneos también en la lista incluyen anemia de diamante-Blackfan, anemia por deficiencia severa de hierro, enfermedad juvenil de Paget y deficiencia de fosfatasa ácida 17). Se han descrito escleras azules adquiridas en adultos con melanosis ocular, melanoma conjuntival, alcaptonuria y enfermedad de Addison. Se pueden indicar derivaciones apropiadas a evaluaciones pediátricas y ortopédicas para manifestaciones no oculares 18).
El asesoramiento genético para trastornos hereditarios asociados puede ser beneficioso para los pacientes con esclerosa azul y otras manifestaciones de enfermedad sistémica.
La intervención quirúrgica puede estar indicada en casos de adelgazamiento y perforación extremos. El soporte estructural puede ser proporcionado por un injerto escleral preservado o fascia autóloga lata, particularmente en casos que requieren sutura de un dispositivo como un implante de tubo 19). Se deben considerar las investigaciones sistémicas y el tratamiento para abordar la patología subyacente.
Pronóstico de esclerótica azul
El pronóstico varía con la presencia de manifestaciones oculares y sistémicas del trastorno subyacente. Los pacientes con esclerótica azul tienen un mayor riesgo de rotura de globo o perforación escleral intraoperatoria durante la cirugía ocular de rutina. Los trastornos sistémicos también son frecuentes en pacientes como fístula carotídea cavernosa, ruptura arterial,pérdida de audición y fracturas óseas 20).
Referencias