- definitioner og prævalens
- diagnose
- Nccah manifestationer hos kvinder
- manifestationer i barndommen
- manifestationer i ungdomsårene og voksenlivet
- Nccah-styring fra den første Manifestation til voksenlivet
- behandling i barndommen
- ledelse i ungdomsårene
- patienter behandlet siden barndommen
- kvinder med første Manifestation under ungdomsårene eller Hyperandrogene symptomer efter behandlingsophør
- Nccah og reproduktion
- subfertilitet
- Miscarriages
- Fertilitetsplanlægning
- Prenatal behandling af CAH
- behandling under graviditet
- andre særlige problemer
- stresshåndtering i nccah
- Psykobiologiske aspekter i NCCAH
- konklusioner
- Forfatterbidrag
- interessekonflikt Erklæring
definitioner og prævalens
medfødt binyrehyperplasi (CAH) omfatter en familie af autosomale recessive lidelser karakteriseret ved mild til akut nedsat cortisolsyntese på grund af en mangel i en af de fem binyrebarksteroidogene stoffer, der kræves til kortisolproduktion (1, 2). Konventionelt er CAH opdelt i (A) klassisk (CCAH), der præsenterer med saltudslip eller den enkle viriliserende form, der manifesterer sig ved fødslen og/eller i den nyfødte periode, og (b) ikke-klassisk (NCCAH), der repræsenterer en mindre alvorlig form for lidelsen, der mangler køns tvetydighed, er ikke umiddelbart livstruende og præsenterer senere i livet eller forbliver asymptomatisk eller fejldiagnosticeres som en anden sygdom (3). Grænserne på tværs af de forskellige former for CAH er imidlertid ikke strengt definerede, hvilket har tendens til at øge udfordringerne forbundet med denne lidelse. Det tilrådes derfor at betragte CAH som et kontinuum af fænotyper, fra svær til mild eller ellers asymptomatisk (3).
den mest almindelige form for CAH er forårsaget af 21-hydroksylasemangel (21ohd), hvilket resulterer i nedsat eller ingen omdannelse af 17 hydroksiprogesteron (17 OHP) til 11 deoksikortisol og af progesteron til deoksikortikosteron. Blokaden af steroidkonvertering resulterer i øget produktion af androgenprecursorer under CRH-ACTH-stimulering, hvilket fører til biokemisk hyperandrogenisme, præget af forhøjede 17-OHP-niveauer. Den klassiske form af sygdommen forekommer hos 1 ud af 16.000 levende fødsler over hele verden. Det skal dog nævnes, at den opfattede forekomst af sygdommen er relateret til den anvendte screeningsmetode. For eksempel er det rapporteret, at forekomsten af den klassiske form for CAH i Sverige er 1:11.500, når der anvendes en case survey-tilgang, hvilket dog falder til 1:9.800, når hormonel screening anvendes. Ligeledes ligger forekomsten i Frankrig mellem 1:15.472 og 1:23.000 og 1: 10.749 og 1: 11.661, når de forskellige screeningsmetoder anvendes (4).1 ud af 1.000 kaukasiere og mere almindeligt i visse etniske grupper, såsom Ashkenasiske jøder (1:27), latinamerikanere (1: 53), Jugoslavere (1: 62) og italienere (1:300) (5, 6). Ikke desto mindre mangler data om forekomsten af NCCAH baseret på forskellige estimeringsmetoder (case-survey, hormonal screening og molekylær test). NCCAH betragtes som den mest almindelige autosomale recessive endokrine lidelse med en bærefrekvens på 1:25 til 1:10. I NCCAH på grund af 21-OHD estimeres den resterende ensymatiske aktivitet til at være omkring 10-70% (7-9).
diagnose
i sammenligning med diagnosen af den klassiske form af sygdommen, som stilles ved fødslen eller i den nyfødte periode på grund af køns tvetydighed og/eller saltspildende symptomer eller gennem screeningsprogrammer, der anvendes i nogle lande, kan de fleste tilfælde af NCCAH ikke let påvises (4, 10). Derudover forbliver mange individer asymptomatiske i barndommen og ungdommen, har normal reproduktiv funktion og bliver kun opmærksomme på NCCAH på grund af diagnosen af et andet familiemedlem og deraf følgende test (11). Imidlertid, de fleste kvinder med NCCAH søger lægehjælp, når de oplever symptomer på androgenoverskud, og, når klinisk mistanke beder om test, forhøjede basale 17 OHP-niveauer vil mere sandsynligt end ikke pege på en diagnose af nccah.
faktisk anbefaler de kliniske retningslinjer foreslået af Endocrine Society en baseline ikke-stimuleret værdi på 17 OHP som screeningsværktøj til nccah. Morgen 17 OHP niveauer >6 nmol/L i follikelfasen hos menstruerende hunner bør bede om yderligere evaluering, da det er vist, at niveauer over 6 nmol/L fanger 90% af nccah-individer (12, 13). Tilfældige målinger på 17 OHP har ikke vist sig at være nyttige, da disse ofte giver normale niveauer hos patienter med NCCAH, og de er desuden ekstremt høje i lutealfasen hos halvdelen af raske kvinder (13). Men i vores data afledt af 280 personer med sygdommen, seks patienter (2.1%) havde en baseline 17 OHP-værdi < 6 nmol/L (14). Derudover Bidet et al., i en stor kohorte af kvinder med NCCAH verificeret ved molekylære teknikker fandt også basale 17 OHP-værdier lavere end 6 nmol/L hos 8% af de undersøgte forsøgspersoner (15). Endelig baseret på data indsamlet af Speiser et al. Viste 9% af personer med NCCAH 17 OHP-værdier lavere end 2 ng/ml (det svarer til 6nmol/L). Ifølge andre undersøgelser er en baseline værdi på 17 OHP mellem 5,1 og 9 nmol/L tilstrækkelig til diagnosen NCCAH (13, 16, 17). For nylig et niveau af basal 17 OHP af 4.6 nmol / L blev foreslået som en tærskel for ACTH-test for at forudsige NCCAH hos personer med for tidlig adrenarche i barndommen (18).
summen af disse fund og forslag indikerer, at udvælgelsen af patienter, der vil gennemgå en Synacthen-stimuleringstest, skal evalueres fra sag til sag. Et 17 OHP poststimuleringsniveau over 3 nmol/L er påkrævet til diagnosen (19). Heterosygoter, der bærer en CYP21A2-mutation, udviser let forhøjede 17 OHP-niveauer efter ACTH-stimulering, selvom der er overlapning hos upåvirkede personer (9). Imidlertid, som Dacou-Voutetakis et al. har påpeget, at hvis summen af basal-og post-stimulering 17 OHP-værdier overstiger 1,5 nmol/L, er muligheden for heterosygositet usædvanlig høj (20).
en anden vigtig overvejelse vedrører de teknikker, der anvendes til 17 OHP evaluering. 17 OHP-niveauer måles ved en række immunoassaymetoder, men som det for nylig er blevet vist, blev de mest nøjagtige og pålidelige resultater opnået ved implementeringen af kombinationen af væskekromatografi med massespektrometri (LC-MS/MS). Faktisk blev der fundet mange falske positive 17 OHP-målinger, når LC-MS/Ms-målinger blev sammenlignet med standardmetoder (21) ikke desto mindre anvendes sidstnævnte procedurer ikke universelt, i tilfælde af en usikker diagnose vil de give mere præcise resultater. Det skal bemærkes, at screenings-og diagnosetærsklerne for LC-MS/MS kvantificeret 17 OHP endnu ikke er defineret. Disse tærskler er sandsynligvis lavere end dem, der er fastlagt med immunoassays på grund af den forbedrede specificitet af LC-MS/MS, en metode, der er mindre tilbøjelig til krydsreaktivitet og interferenser (22, 23). Hvorvidt der kræves en urinsteroidprofil til den endelige diagnose, skal stadig belyses (22). I grænsetilfælde anbefales det at opnå en komplet adrenokortikal profil efter ACTH-stimuleringstesten for at differentiere 21-hydroksylasemangel fra andre defekter og etablere en fast diagnose.
specifikt bør en komplet steroidprofil udføres i tvetydige tilfælde for at bekræfte 21-hydroksylasemangel og udelukke andre detekterer. Inklusion af 17 OHP, cortisol, 11-deoksikortikosteron,11-deoksikortisol,17-OH-pregnenolon, Dehydroepiandrosteron og androstenedion i en serumsteroidprofil ville være nyttigt til at udelukke andre årsager til Cah, såsom 11-HH-mangel og, mere sjældent, 3-HH-HH-mangel eller P450-HH-mangel (24). Mens genetisk testning ikke anses for at være et primært diagnostisk værktøj til NCCAH på dette tidspunkt, er det obligatorisk for diagnosebekræftelse og for genetisk rådgivning (3).
for så vidt angår cortisolniveauerne forventes generelt en poststimuleringsværdi på mindst 496 nmol/L. Af note, ifølge Stoupa et al., 60% af 47 børn med NCCAH som et resultat af 21 OHD havde lave cortisolværdier efter stimuleringen, et fund, der pegede på behovet for øget overvågning for udvikling af binyreinsufficiens under større stressorhændelser (25).
Nccah manifestationer hos kvinder
den kliniske ekspression af NCCAH er kendetegnet ved et højt niveau af polymorfisme med hensyn til ikke kun begyndelsesalder, men også de forskellige tegn og symptomer. Det rapporteres, at den første kliniske præsentation af NCCAH er i 11% af tilfældene før 10 år og i 80% mellem 10 og 40 år (12). Genotype-fænotype-korrelationen I CAH og NCCAH er endnu ikke belyst. Speiser et al. antyder, at de fleste, men ikke alle, fænotypiske variationer i 21-hydroksylasemangel skyldes allel variation i CYP21A2 (26).
manifestationer i barndommen
i den nyfødte periode forbliver typisk nccah-hunner asymptomatiske og har normale ydre kønsorganer. Det tidligste tilfælde af nccah rapporteret er en 6 måneder gammel pige, der udviklede skamhår (11). Normalt starter kliniske fund og symptomer i nccah-tilfælde fra en alder af 5 år eller endda senere og er relateret til øgede androgenniveauer, selvom mild kortisolmangel også kan forekomme i nogle tilfælde (8). I 92% af nccah-tilfældene er det første symptom, der manifesterer, for tidlig pubarche (PP), der forekommer under 8 år hos piger og under 9 år hos drenge (12) med en estimeret prævalens på mellem 10 og 11,3% (15). Det bemærkes, at forekomsten af NCCAH varierer fra 5 til 30% (20, 27) hos børn, der henvises til PP. Baseret på vores Analyse af 280 patienter med molekylær bekræftelse af NCCAH fandt vi imidlertid, at forekomsten af PP hos 94 kvinder yngre end 8 år var så meget som 88%. På den anden side identificerede en analyse af 45 mænd med NCCAH PP hos kun 29% af forsøgspersonerne (14). Desuden skal vi huske på, at omkring halvdelen af personer med PP kan være heterosygotbærere af en CYP21A2-mutation.
et andet aspekt, der skal afklares, er forholdet mellem PP og ældre pubertet. Det antages, at den perifere aromatisering af adrenale androgener til østrogener i nogle tilfælde kan aktivere hypothalamus-hypofyse-gonadalaksen, hvilket fører til sekundær for tidlig pubertet (28). I et sådant tilfælde er der behov for regelmæssig opfølgning. Nogle børn udvikler hirsutisme, akne og/eller hurtig vækst og manifesterer høj struktur og udvikling af knoglealder (29). Sidstnævnte kan resultere i afkortet sluthøjde som en konsekvens af hurtig epifysisk fusion. Andre sjældne kliniske fund i barndommen inkluderer labial vedhæftning, perianalt hår, klitoromegali og stemmehæshed (30-32).
manifestationer i ungdomsårene og voksenlivet
under ungdomsårene og voksenalderen præsenterer NCCAH hos kvinder hyperandrogene symptomer, der ligner polycystisk ovariesyndrom (PCOS) (33). Symptomer inkluderer hirsutisme, acne, androgen alopeci, anovulation, menstruationsdysfunktion og infertilitet. I en multicenterundersøgelse var de mest almindelige symptomer blandt unge og voksne kvinder hirsutisme (59%), oligomenorrhea (54%) og acne (33%). Blandt 161 kvinder med NCCAH var symptomer hirsutisme (78%), menstruationsdysfunktion (54,7%) og nedsat fertilitet (12%) (34). I vores gruppe på 161 kvinder med sygdommen præsenterede 63% af patienten en polycystisk ovarielignende fænotype (14). Dette fund har en omvendt implikation, da forekomsten af NCCAH er høj hos kvinder med hyperandrogenæmi. Sikkert, undersøgelsen af Vitcel et al. rapportering af en forekomst af NCCAH fra 1 Til 33% hos hyperandrogene kvinder er af stor interesse. I en anden undersøgelse af 950 kvinder, der blev henvist til evaluering af androgenoverskud, blev NCCAH påvist hos 4,2% af dem. Imidlertid var nccah-patienterne yngre og mere hirsute sammenlignet med de andre grupper af hyperandrogene patienter. De blev også karakteriseret ved signifikant højere niveauer af testosteron, frit testosteron og 17 OHP end patienter med andre hyperandrogene syndromer. I ovarie ultralyd viste 77% af dem polycystiske æggestokke og 41% øget ovariestørrelse. Samlet set opfyldte de PCOS-kriterierne i henhold til NIH og Rotterdam med en procentdel på henholdsvis 56 og 72,8% (35).
sygdommens progressive karakter fremhæves af det faktum, at forekomsten af hirsutisme har vist sig at stige med alderen og er blevet observeret at være sjælden før puberteten. Hirsutisme er den mest almindelige kliniske manifestation rapporteret hos patienter med NCCAH, der spænder fra 71 til 96% (28, 36). Pubertal piger med NCCAH typisk til stede med hirsutisme (37, 38). I den anden ende af spektret er spørgsmålet om alopeci. Skaldethed hos mænd er rapporteret hos nccah-patienter. Udbredelsen af alopeci syntes også at stige med alderen, fra 6% hos patienter i det andet årti af deres liv til 19% i det femte, hvilket igen indikerer sygdommens progressive karakter (39). Acne er rapporteret i næsten 33% af nccah forsøgspersoner (36). Bemærkelsesværdigt blev mutationer af CYP21A2-genet, der forårsager NCCAH, påvist hos 4,9% af 123 voksne kvinder med svær acne (40).
menstruelle uregelmæssigheder inklusive oligomenorrhea eller sekundær amenorrhea kan ofte være det præsenterende tegn på NCCAH hos personer efter menarche. Derudover oplever 8-9% af tilfældene også primær amenorrhea og søger derfor lægehjælp for første gang. Desuden er oligomenorrhea blandt nccah-individer mere almindelig end primær amenorrhea. For eksempel i en serie fra Moran et al. blandt NCCAH-kvinder i alderen 10 til 19 år var oligomenorrhea til stede i 56% af tilfældene sammenlignet med kun 9% af ungdomsårene med primær amenorrhea (12). Afslutningsvis ligner så mange nccah-symptomer PCOS, at det ikke er overraskende, at NCCAH er blevet kaldt den store mimicker af PCOS. Under alle omstændigheder bør en diagnose af NCCAH overvejes under evalueringen af enhver ung kvinde, der henvises til hyperandrogene symptomer.
i en undersøgelse foretaget af Arlt et al. blev patienter med NCCAH karakteriseret ved signifikant højere BMI og lavere sluthøjde sammenlignet med matchede kontroller (41). Disse data i forbindelse med nogle få undersøgelser, der indikerer tilstedeværelsen af insulinfølsomhed, antyder en ugunstig metabolisk profil, der kunne patofysiologisk forklares ved effekten af forhøjede androgenkoncentrationer og/eller som et resultat af glukokortikoidbehandling (42, 43). Osteoporose kan også påvises hos disse forsøgspersoner, sandsynligvis som en konsekvens af kortikosteroidbehandlingen (34).
Nccah-styring fra den første Manifestation til voksenlivet
Når diagnosen NCCAH er etableret, berettiger flere spørgsmål relateret til efterfølgende behandlingsanbefalinger overvejelse. Det første spørgsmål, der skal behandles, er, om glukokortikoidbehandling (GC) er indiceret. Den generelle begrundelse bag denne tilgang er, at ved at tilvejebringe tilstrækkelige cortisolkoncentrationer til patienten dækkes hendes daglige behov, og følgelig vil CRH-ACTH-aksestimulering være tilspidset, hvilket fører til nedsat adrenal androgenproduktion. Den generelle regel vedrørende denne tilgang er, at GCs gives ved erstatning og ikke farmakologiske doser, mens indflydelsen af alder, køn, laboratoriedata, patientspecifikke anbefalinger og mål for behandling på glukokortikoidudskiftningsterapi tages alvorligt i betragtning. Vi skal dog også være opmærksomme på, at langvarig GC-substitutionsbehandling kan føre til hyperkortisolæmi med de resulterende veldokumenterede bivirkninger på alle aspekter af stofskiftet, især vækst, fedtfordeling og insulinresistens såvel som på psykologisk profil. En anden stor ulempe ved denne tilgang er manglen på et tilstrækkeligt klinisk indeks eller biokemisk markør for tilstrækkelig udskiftningsdosis, såsom findes med hensyn til TSH-værdier i hypothyroidisme.
endelig komplicerer det faktum, at der ikke er enighed om, hvilken GC der skal anvendes, og fraværet af langsigtede data vedrørende forskellige former for GC-administration yderligere behandlende lægeres beslutninger om og valg af det optimale valg for hver patient. Hydrocortison anvendes typisk til børn, da det mest ligner det naturlige hormon (cortisol), men det betragtes ikke som en passende tilgang hos unge og unge kvinder på grund af behovet for flere daglige doser. Derfor foretrækker de fleste voksne endokrinologer enten deksamethason eller prednisolon ved passende doser. På den anden side skal det påpeges, at ækvivalensen af forskellige GCs er baseret på deres antiinflammatoriske virkning og ikke på forskellige aspekter af menneskelig metabolisme. Da cortisol påvirker næsten 20% af det humane genom, forventes forskellige reaktioner af forskellige GCs i forskellige væv. Det bekræfter denne opfattelse, at administration af deksamethason til patienter med CAH har vist forringelse af insulinresistensindekserne sammenlignet med andre GCs (44). Derudover udøver administrationen af 2,5–7,5 mg prednisolon, en dosis, der betragtes som normal, en langvarig negativ indvirkning på knoglemetabolismen (45). Derfor bør hydrocortison betragtes som den bedste form for behandling i tilfælde af GC-tilskudsterapi. Fremkomsten af den nyligt syntetiserede hydrocortisonformel med en pille om dagen og dens første positive resultater hos patienter med CAH viser meget løfte for fremtiden (46).
behandling i barndommen
nccah-patienter, der diagnosticeres i barndommen med tegn på PP, kan behandles med hydrocortison med det formål at undertrykke binyrehormonerne og forhindre hurtig udvikling af knoglealder, der kan påvirke den endelige højde. Hos de patienter, hvor hydrocortisonbehandling blev påbegyndt 1 år før pubertetens begyndelse, og som havde en knoglealder under 9 år, forblev den endelige højde inden for det genetiske potentiale (47). Tilgængelige undersøgelser indikerer, at voksenhøjde nærmede sig den forventede målhøjde hos patienter, der blev nøje overvåget, og som strengt overholdt medicinplaner (48-50). En nylig lille undersøgelse af fem tilfælde (patienter i alderen 6,1–9,2 år) viste, at en ultralav dosis er en lovende mulighed for at reducere endogen stress og dens virkninger. Hvorvidt dette udgør en realistisk tilgang, og hvor meget den administrerede dosis skal være, skal stadig belyses (51). En undersøgelse foretaget af Nebesio et al. hos 9 præpubertale patienter med CCAH sammenlignede de hormonelle virkninger og farmakokinetikken af hydrocortison, prednisolon og deksamethason. De viste, at deksamethason var mere potent end de andre former for at opnå signifikant lavere binyrehormonniveauer, hvilket tyder på, at deksamethason er mere effektivt til undertrykkelse af adrenal androgenproduktion (46). Naturligvis er der behov for yderligere undersøgelser for at verificere eller afvise dette fund. Desuden kan forhøjede androgenkoncentrationer i nogle tilfælde føre til sekundær stimulering af GnRH-aksen, hvilket fører til for tidlig pubertet. I et sådant tilfælde kan et parallelt forløb med GnRH-analog ordineres, hvis knoglealderen er signifikant højere end kronologisk alder og/eller projiceret sluthøjde er uforholdsmæssig i forhold til målhøjden.
en anden vigtig overvejelse vedrører tilfælde af samtidig GH-mangel i tilfælde af, at behandling med GnRH-analoger og GH-recept resulterede i opnåelse af målhøjde (52). Ifølge de nuværende retningslinjer anbefales den ovenfor beskrevne terapi kun forsigtigt i de tilfælde, hvor den forudsagte højde SD er -2,25 < målhøjden eller endnu lavere (53). En alternativ indikation for start af hydrocortisonbehandling er en utilstrækkelig cortisolrespons efter ACTH-stimulering (36).
den foretrukne GC-behandling hos børn er normalt hydrocortison 10-15 mg/m2, opdelt i tre doser. Ofte har lavere doser også vist sig effektive, startende fra 6 mg/m2 / dag (34). Overdosering bør undgås, i betragtning af det, der kan resultere i dårlig vækst samt Cushingoid funktioner. Regelmæssigt vækstmønster, en knoglealder, der er kompatibel med kronologisk alder, og fravær af central fedme kan også tjene som kliniske indekser for passende styring. Med hensyn til biokemisk / hormonel profil betragtes androstenedion-og testosteronniveauer i midten til øvre intervaller for knoglealder som bedre markører for tilstrækkelig GC-erstatningsterapi hos børn med NCCAH. Imidlertid blev undertrykte testosteronniveauer fundet hos 10% af nccah-patienterne, mens yderligere 28% af patienterne havde øget testosteronkoncentrationer, hvilket muligvis kan tilskrives hydrocortisonvariabilitet (54, 55). DHEAS-værdier nedsættes med meget små doser, mens undertrykkelsen af 17 OHP-og progesteronniveauer kræver meget høje GC-doser. Det skal bemærkes, at 17 OHP–værdier er afgørende for diagnosen, men ikke nyttige under opfølgningen. Bemærk, at blodprøveudtagning til hormonel evaluering skal udføres uden ophør af behandlingen. På grund af sygdommens forvirring og dens mangesidede natur er der imidlertid ingen specifikke retningslinjer for tidspunktet for regimeændringer eller ophør af glukokortikoidbehandling hos børn.
ledelse i ungdomsårene
patienter behandlet siden barndommen
indtil etableringen af det normale menstruationsmønster hos nccah-piger anbefales fortsættelsen af GCs, der startede i barndommen (56). Imidlertid viser unge patienter ofte ikke tilstrækkelig overholdelse af kronisk administration af lægemidler og udelader ofte doser. Derudover falder halveringstiden for hydrocortison i puberteten med 50% som et resultat af øgede IGF-1-niveauer, hvilket formindsker 11-prisohsd-aktivitet såvel som på grund af øget cortisolclearance som følge af amplifikation af glomerulær filtreringshastighed (57). I den unge kvinde, 2 år efter menarche, og hvis der er registreret normale ovulatoriske cyklusser, anbefales en patientcentreret tilgang til de hyperandrogene symptomer, der kan forekomme.
hvis der er alvorlige hyperandrogene fund, såsom hirsutisme, acne og/eller oligomenorrhea, vil fortsættelse af behandlingen blive overvejet. Det er vigtigt at huske den skrøbelige følsomhed hos en ung ungdom, som vil blive alvorligt beskadiget af de “afstødende” tegn på hyperandrogenisme. Kombinationen af hyperandrogene tegn, menstruationsforstyrrelser og dårlig livskvalitet er veldokumenteret, og den er særlig høj hos yngre kvinder. Et andet punkt, der skal overvejes, er responsen fra adrenal reserve efter ACTH-stimulering. Da peak cortisol hos pubertal og voksne kvinder efter stimulering er under 496 nmol / L, bør steroidbehandling administreres i tilfælde af stress (58). På den anden side, hvis ingen af ovenstående symptomer opstår hos den unge patient, kan GC-behandling afbrydes, hvorefter regelmæssig opfølgning anbefales.
kvinder med første Manifestation under ungdomsårene eller Hyperandrogene symptomer efter behandlingsophør
i nærvær af hyperandrogene symptomer anbefales en patientorienteret tilgang stærkt med fokus på patientens hovedklager. Dette er langt bedre end en generel universel Modus operandi, da den unge patient med sidstnævnte tilgang vil blive skuffet, hvilket fører til seponering af behandlingen og som et resultat til ugunstige sundhedsresultater i voksen alder. I tilfælde med svær hirsutisme udgør et kursus på 6 til 12 måneder med orale p-piller (OCPs) en rimelig procedure i betragtning af de gavnlige virkninger af OCPs på SHBG-leverproduktion og faldet i androgenfrigivelse fra æggestokken. Klinisk forbedring forventes efter mindst 2-3 måneders OCPs-initiering, mens samtidig brug af en progestagen med minimale androgene egenskaber anbefales stærkt. Hvis OCP ‘ er mislykkes som første linje tilgang, kan antiandrogener (spironolacton, flutamid, bicalutamid, cyproteronacetat og finasterid) tilsættes til behandlingen. Efter vores erfaring har administrationen af bicalutamid opnået signifikant forbedring i tilfælde af svær acne, men lignende resultater blev ikke opnået i tilfælde med alvorlig hirsutisme. Kosmetiske tilgange såsom laserapplikation og hårfjerningsmidler kan også foreslås for kvinder, der klager over overdreven eller uønsket hårvækst eller hårtab i hovedbunden (androgen alopeci) (32, 34, 57).
hos patienter med utilstrækkelig cortisolsekretion efter stimulering, eller hvis OCPs og antiandrogener ikke kan tolereres, anbefales et kursus med GCs (57). Data fra Ny et al. Angiv, at uregelmæssige menstruationer og acne vendes inden for 3 måneder efter indledningen af glukokortikoidbehandlingen, mens hirsutisme kræver næsten 30 måneder (59). I modsætning til barndommen anvendes der i ungdomsårene ofte langtidsvirkende steroider, og der anbefales regimer på 5 mg prednisolon eller 0,25 mg deksamethason (53). Imidlertid har kliniske data i den virkelige verden vist en række forskellige regimer anvendt i nccah-ledelse (41).
det primære mål med behandlingen bør være patientens lindring fra de hyperandrogene symptomer. Således blandt personer med nccah diagnosticeret udelukkende baseret på laboratorieresultater, kliniske træk bør vejlede ledelsesanbefalinger, da hormonelle og molekylære fund ikke nødvendigvis forudsiger klinisk sværhedsgrad. I henhold til Endocrine Society-retningslinjerne bør nccah-patienter have mulighed for at afbryde GC-behandling, når symptomerne forsvinder (60). Naturligvis bør disse patienter ikke gå tabt for opfølgning, mens behandlingen skal genoptages i tilfælde af gentagelse af symptomerne. I tilfælde af seponering skal patienterne informeres om muligheden for infertilitet og bør opfordres til at søge lægehjælp, hvis de ønsker at blive gravid (19). Bemærk, at den passende overførsel af patienten fra den pædiatriske til den voksne endokrinolog skal udføres optimalt efter 1 års synkroniseret overvågning (53, 61).
Nccah og reproduktion
subfertilitet
subfertilitet rapporteres almindeligvis i klassiske former for CAH, hvilket hovedsageligt skyldes menstruationsforstyrrelser, kronisk anovulation og anatomiske deformiteter. Fødselsraten er estimeret til 17% sammenlignet med 65% af kontrolpopulationen (62). I mellemtiden er data vedrørende fertilitet hos kvinder med NCCAH for nylig blevet vurderet detaljeret, og den estimerede infertilitetsforekomst er 11% blandt nccah-kvinder, det vil sige relativt mildere end I CAH, og er i mange tilfælde let løst. Bidet et al. evalueret fertilitet hos 190 kvinder, der lider af NCCAH, hvoraf 95 ønskede at blive gravid. I denne population blev der rapporteret 187 graviditeter hos 85 kvinder, hvilket resulterede i 141 fødsler hos 82 personer. Det skal fremhæves, at 99 af graviditeterne (52, 9%) forekom før diagnosen NCCAH, tre af dem med anvendelse af ægløsningsinduktionsprotokoller, resten var spontan, mens 88 graviditeter fandt sted efter nccah-diagnosen. I langt de fleste af dem udviklede graviditeten sig efter behandlingsinstitutionen med hydrocortison, mens det i 11 kvinder skete spontant (63, 64). I en anden rapport af 22 observerede nccah kvinder, der ønsker graviditet, 12 graviditeter fulgte med prednison (65).
Miscarriages
i kohorten af Bidet et al. var abortfrekvensen 6, 5 og 26, 3% hos patienter behandlet med henholdsvis GCs og ubehandlede patienter (64). Resultatet af graviditeten kan endda være vellykket uden nogen glukokortikoidbehandling i tilfælde, hvor NCCAH endnu ikke var diagnosticeret, som også rapporteret i en sagsrapport fra Cuhaci et al. Ikke desto mindre oplevede det samme par to aborter og rapporterede subfertilitet efter denne første graviditet (66). Hvad angår muligheden for for tidlige graviditeter, som vurderet af Bidet et al., det adskiller sig ikke væsentligt fra resten af befolkningen (15).
Fertilitetsplanlægning
for de kvinder med symptomatisk hyperandrogenisme eller med rapporteret infertilitet, men som ønsker at blive gravid, anbefales GC-terapi stærkt. I denne periode er de anvendte glukokortikoider prednison eller hydrocortison. Den vigtigste handling, fordi den vil lede de efterfølgende trin, er genetisk test af de potentielle forældre. Kvinder med NCCAH, der ønsker at blive gravid, bør være opmærksomme på risikoen for at føde et spædbarn med den klassiske form af sygdommen. Resultatet af graviditeter blandt kvinder med NCCAH, og mere specifikt forekomsten af spædbørn født med CCAH, anslås i nylige undersøgelser at være mellem 1, 5 og 2, 5%, med NCCAH omkring 15% (36, 64). Selvfølgelig afhænger denne frekvens af referencepopulationen, en højere forekomst forekommer i populationer med høje blandede ægteskaber (36). De forventede chancer for, at forældre føder et barn med CAH, er 1 ud af 120 for ukendt faderlig genotype, nul, hvis faderen ikke har nogen relevant mutation, og 1 ud af 4, hvis han har en heterosygøs mutation (19). Som følge heraf er genetisk testning af disse kvinders partnere afgørende for at vurdere risikoen for at føde et barn med den klassiske form for CAH (64, 67).i tilfælde af at den potentielle mor bærer to NC-mutationer (tabel 1), er der ikke noget absolut behov for genetisk testning i den fremtidige far, da hvis han er heterosygøs for CYP12A2-mutationerne, risikerer afkomene at udvikle den ikke-klassiske form af sygdommen i fremtiden. Det skal dog bemærkes, at bærerstatus er 2-15% i forskellige populationer, og halvdelen af disse individer bærer en alvorlig mutation. Desuden er endelige anbefalinger vedrørende situationer, hvor genetisk test ikke er påkrævet, vanskelige i betragtning af den ufuldkomne genotype-fænotype-korrelation, især for mildere mutationer . For eksempel forekommer p30l-mutationen, hyppigst forbundet med NCCAH, også hos patienter med klassisk Cah . En stor europæisk multicenter-undersøgelse viste for nylig, at dette var tilfældet hos 78% af patienterne . I denne undersøgelse var den milde v218l-mutation forbundet med klassisk snarere end NCCAH i 30% af tilfældene. Derfor bør alle patienter med NCCAH tilbydes genetisk rådgivning og molekylær vurdering af reproduktive partnere.
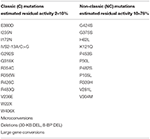
tabel 1. Mutationer af CYP21A2 med minimal (C) eller moderat (NC) restaktivitet (14, 68, 69).i modsætning hertil, hvis den potentielle mor er en sammensat heterosygot med en alvorlig (C) og en NC-mutation, er genetisk testning af den fremtidige far obligatorisk. Vi skal huske på, at data, der stammer fra undersøgelser, der bruger immunoassays eller de mere nøjagtige LC-MS/MS-procedurer, konsekvent fremhæver manglende evne til pålideligt at udelukke heterosygositet ved hjælp af basale eller ACTH-stimulerede 17 OHP-værdier på grund af den betydelige overlapning med det normale interval. Man kan foreslå brugen af en Synacthen-test, og hvis den ikke er kompatibel med heterosygositet (summen af basale og peak stimulerede 17 OHP-værdier < 1,5 nmol/L), kunne DNA-test undgås. Vi bør dog være meget forsigtige med fortolkningen af denne test, da den ikke er blevet valideret i andre undersøgelser. Derfor kan brugen af ACTH-stimuleret 21-deoksikortisol enten enkeltvis eller som en del af et steroidforhold eller steroidprofil lette den biokemiske identifikation af heterosygoter i fremtiden, især da LC-MS/MS bliver mere bredt tilgængelig . Hvis den potentielle far bærer en NC-mutation, er der ikke behov for noget andet. Omvendt, hvis han er bærer af en klassisk mutation, vil spørgsmålet om prænatal behandling af fosteret opstå. Alle mulige genotypekombinationer og de foreslåede procedurer er vist i tabel 2.

tabel 2. Planlægning af foreslåede procedurer under graviditet baseret på potentielle forældre genotype.
Prenatal behandling af CAH
formålet med prenatal behandling af CAH er forebyggelse af genital virilisering af fosteret, men også lindring af stress følt af forældrene, der sandsynligvis vil have et barn med tvetydige kønsorganer (70). Det anvendes på grund af dets evne til at krydse placenta, og fordi det ikke deaktiveres fra placenta 11-cholagogue-steroiddehydrogenasen og kun binder minimalt til moderens blodkortisolbindende globulin (CBG). Ved at undertrykke føtal ACTH-sekretion nedsætter den forhøjede føtal androgenproduktion. Behandlingen skal afbrydes i tilfælde af, at karyotype-eller DNA-analyse afslører henholdsvis en mand eller en upåvirket kvinde (71). Ikke desto mindre, selvom føtal genital virilisering allerede er startet 6-7 uger efter undfangelsen, kan chorioniske villøse biopsier til genetisk diagnose ikke opnås før den 10.-12. uge. Dette tidsinterval antyder, at alle graviditeter, der er i fare for virilisering af CAH, skal behandles, selvom kun 1 ud af 8 fostre er påvirket og kvindelige (19, 34, 70).
hvorvidt denne eksponering af fosteret for deksamethason til forebyggende foranstaltninger er medicinsk og etisk acceptabel forbliver kontroversiel (70). Dyreforsøg har vist, at første trimester nedsætter fødselsvægten, påvirker nyrepankreatisk betacellefunktion og hjerneudvikling og disponerer for angst, hypertension og hyperglykæmi i voksenalderen (70). Desuden klager 10-20% af gravide kvinder, der bruger deksamethasonbehandling under graviditet, over vægtøgning, øget appetit, humørsvingninger, søvnløshed, ødemer, hypertension, hyperglykæmi og striae. Imidlertid, nyt et al. Prader-score var lavere end gennemsnittet hos fostre behandlet med prenatalt, men ingen forskel i langtidsresultater (72). Derudover, i en anmeldelse af Merce Fernandes-Balsells et al. var forbundet med reduktion i føtal virilisering uden signifikante maternelle eller føtale bivirkninger (63). Forfatterne af ovennævnte gennemgang samt de på hinanden følgende retningslinjer for det endokrine samfund påpeger imidlertid, at emnet på grund af de små stikprøvestørrelser i hele litteraturens krop forbliver usikkert, og yderligere undersøgelse er klart nødvendig.
beslutningen om at indlede behandling bør kun foretages i store centre med et erfarent team og protokoller godkendt af institutionelle Gennemgangsudvalg og baseret på familiens værdier og præferencer og med deres skriftlige informerede samtykke som en forudsætning (19, 63). Flere specialister på området indikerer, at der bør lægges en højere værdi på at forhindre unødvendig prænatal eksponering af mor og foster for deksamethason snarere end at pålægge forældre og patienter den følelsesmæssige vejafgift af tvetydige kønsorganer. Vigtigst er det, at det bør præciseres over for forældrene, at Administration ikke ændrer patientstatus, men er rettet mod at reducere behovet for operation snarere end at bevare liv eller intellektuel kapacitet. De skal også vide, at sandsynligheden for, at deres barn vil lide af den klassiske form af sygdommen, er høj, på trods af behandling. I mellemtiden er eksponeringen af en ung organisme for en meget potent GC i en særlig følsom periode med føtalprogrammering og vækst, som meget vel kan vise sig ubrugelig i tilfælde af et foster med KSY-karyotypen, understøttes ikke på nuværende tidspunkt af robuste og ubestridelige data.
ideelt set bør der være en tidlig diagnose, dette udføres via en ikke-invasiv teknik og inden begyndelsen af fostrets genital virilisering. Arbejde i denne retning peger nye undersøgelser på brugen af cellefrit føtal DNA opnået fra moderplasma som en lovende metode, der muliggør bestemmelse af føtal køn og diagnose af CAH i en tidlig svangerskabsalder (<9 uger). Hvis denne procedure er bredt implementeret i klinisk praksis, undgås unødvendig prænatal deksamethasonbehandling (73).
behandling under graviditet
under graviditet bør kvinder med NCCAH fortrinsvis behandles med hydrocortison, ikke kun fordi graviditet typisk ledsages af forhøjede stressniveauer, men også som en forebyggende foranstaltning mod ovennævnte høje forekomst af aborter. Denne form for glukokortikoid er begunstiget på grund af dets metabolisering ved hjælp af placenta11-cholagogue Oh steroiddehydrogenase II, som forhindrer glukokortikoid i at have nogen virkning på fosteret (71). På undfangelsestidspunktet bør hydrocortisondosis øges til 20-25 mg/dag, og dosisændringer udføres hver 6-8 uge med det formål at holde testosteronniveauet på de øvre normale niveauer i graviditetstrimesteren (74). Som et eksempel vises testosteron-og D4-værdierne hos en patient med NCCAH fra undfangelse op til 3 år efter levering af to sunde tvillinger (Figur 1).
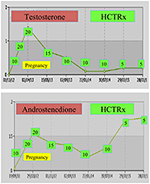
Figur 1. Testosteron og andostenedion niveauer og efterfølgende hydrocortison dosering, fra undfangelsen indtil 3 år efter fødslen administreret til en patient med NCCAH.
andre særlige problemer
stresshåndtering i nccah
under større livstruende stress, kirurgi eller alvorlig sygdom kan patienter med nccah, der behandles med glukokortikoid, kræve større eller hyppigere doser glukokortikoider, da deres binyrefunktion er iatrogenisk undertrykt. Det er derfor afgørende at uddanne forældre til små børn samt at genuddanne patienter om deres overgang til voksenpleje om stressdosering. For nccah-patienter, der ikke behandles med GCs eller i tilfælde af seponering, bør cortisolrespons på ACTH vurderes. Næsten en tredjedel af nccah-patienterne reagerer utilstrækkeligt på stimuleringen (15, 75). For dem, der reagerer normalt på ACTH-stimulering, anbefales ingen behandling med stressdoser (19). Mineralocorticoidbehandling er ikke påkrævet i nogen af tilfældene, da disse patienter har normal aldosteronsekretion og ikke udvikler saltudslip (58). Det bemærkes, at i tilfælde af hyperthyroidisme eller en stigning i behandling med levothyroksin øges cortisolclearance, og der kan forekomme en binyrekrise (58, 76).
Psykobiologiske aspekter i NCCAH
Meyer-Bahlburg et al. har i flere undersøgelser rapporteret nedsat psykologisk profil blandt patienter med NCCAH på grund af 21oh-mangel. Mere specifikt havde de berørte kvinder en højere maskulinisering / defeminiseringsscore på flere målinger af kønsrelateret adfærd sammenlignet med normale kontrolkvinder, skønt markant mindre end hos kvinder med klassisk CAH. De havde også signifikant øget biseksuel eller homoseksuel orientering. Retrospektive klinisk-kvalitative samtaler med disse kvinder afslørede en historie med ubehag og social stress relateret til deres forbehandlingsoplevelser med androgenafhængige tegn, såsom acne, hirsutisme og undfangelsesvanskeligheder (59, 77, 78). Tilsvarende, Arlt et al. viste, at subjektiv sundhedsstatus var signifikant nedsat på tværs af alle otte domæner i en kortvarig sundhedsundersøgelse, med de mest fremtrædende forskelle, sammenlignet med alders-og kønsmatchede kontroller, vedrørende domænerne generel sundhed, vitalitet, og rollebegrænsninger på grund af følelsesmæssige problemer. Hospital angst og Depression Scale (HADS) spørgeskema afslørede også øget angst score (41). Under hensyntagen til disse fund bør psykologiske parametre til vejledning af terapi overvejes hos kvinder med NCCAH, og i forbindelse med den patientorienterede tilgang skal der tilbydes en psykologisk diagnose og støtte.
konklusioner
NCCAH betragtes som den hyppigere og mildere form for CAH på grund af tilbageholdelse af 20-50% f.eks. De fleste patienter kan søge lægehjælp på ethvert tidspunkt i deres liv på grund af symptomer relateret til androgenoverskud. Disse omfatter for tidlig pubarche, hurtig vækst, hirsutisme, acne, menstruelle uregelmæssigheder eller infertilitet samt mange andre mindre fremtrædende manifestationer. Tidlig morgen baseline værdier på 17 OHP som en god indledende screeningstest og yderligere evaluering med ACTH-stimulering og, i tilfælde af grænseresultater, genetisk test, anbefales. Som en generel regel bør androstenedion og ikke 17 OHP niveauer anvendes under opfølgningen.
behandlingsbeslutningen bør baseres på vurdering af fakta og bør følge en individualiseret tilgang. Behandling er ikke altid indiceret, medmindre patienten er symptomatisk, for eksempel børn med tidlig begyndelse og hurtig progression af skam-og kropshår, hurtig vækst og/eller skeletfremgang eller kvinder med oligomenorrhea, acne, hirsutisme, infertilitet eller en kombination af disse og andre af de ovennævnte symptomer. Genetisk rådgivning anbefales kraftigt i nccah kvinder, der ønsker at blive gravide, samt genotypebestemmelse af faderen. Den overordnede styring af patienten inkluderer desuden styring af de sandsynlige komplikationer ved glukokortikoidbehandling eller stofskifterelaterede manifestationer af sygdommen. Endelig er det meget vigtigt for den behandlende læge at holde sig ajour med al udvikling på dette område og integrere de nye data i hans kliniske praksis, da mange terapeutiske problemer relateret til den passende behandling af disse patienter endnu ikke er belyst. Det er helt sikkert nødvendigt med yderligere kliniske undersøgelser på dette område.
for at opsummere, for nccah-kvinden er den ideelle tilgang en skræddersyet, der indeholder en jævn overgang af hendes ledelse, når hun først er henvist fra pædiatrisk til voksen endokrinolog sammen med symptomorienteret behandling, der vil ledsage hende gennem hele sit liv. I betragtning af de mange faktorer, der påvirker det hyperandrogene system, tilrådes det at tilskynde patienter til at vedtage en sundere livsstil ved at forbedre deres kostvaner, øge træningen og sigte mod vægttab. Desuden er psykologisk støtte i visse tilfælde ofte gavnlig. Desuden har specialister inden for områder, der er involveret i behandlingen af denne lidelse, såsom hudlæger, gynækologer og psykologer, brug for en dybdegående forståelse af håndteringen af mistænkelige eller allerede diagnosticerede tilfælde af nccah. Lad os afslutningsvis huske på det indsigtsfulde citat fra den amerikanske astronom Vera Cooper Rubin: “videnskaben skrider bedst frem, når observationer tvinger os til at ændre vores forudfattede meninger.”
Forfatterbidrag
alle forfattere, der er anført, har ydet et væsentligt, direkte og Intellektuelt Bidrag til Værket og godkendt det til offentliggørelse.
interessekonflikt Erklæring
forfatterne erklærer, at forskningen blev udført i mangel af kommercielle eller økonomiske forhold, der kunne fortolkes som en potentiel interessekonflikt.
1. Hvid PC. Medfødt adrenal hyperplasi. N Engl J Med. (2003) 349:776–88. doi: 10.1056 / NEJMra021561
PubMed abstrakt | CrossRef Fuld tekst | Google Scholar
2. Turcu af, Auchus RJ. Adrenal steroidogenesis and congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. (2015) 44:275–96. doi: 10.1016/j.ecl.2015.02.002
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Speiser PW. Nonclassic adrenal hyperplasia. Rev Endocr Metab Disord. (2009) 10:77–82. doi: 10.1007/s11154-008-9097-x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Gidlöf S, Falhammar H, Thilén A, von Döbeln U, Ritzén M, Wedell A, et al. Hundrede års medfødt adrenal hyperplasi i Sverige: En retrospektiv, befolkningsbaseret kohortestudie. Lancet Diabetes Endocrinol. (2013) 1:35–42. doi: 10.1016/S2213-8587(13)70007-
PubMed abstrakt | CrossRef Fuld tekst | Google Scholar
5. P, Dupont B, Rubinstein P, pladsen A, Kastelan A, nye MI. Høj frekvens af ikke-klassisk steroid 21-hydroksylase mangel. Am J Hum Genet. (1985) 37:650–67.
PubMed Abstract | Google Scholar
6. Wilson RC, Nimkarn S, Dumic M, Obeid J, Azar MR, Azar M, et al. Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Mol Genet Metab. (2007) 90:414–21. doi: 10.1016/j.ymgme.2006.12.005
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Miller WL. Clinical review 54: genetics, diagnosis, and management of 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1994) 78:241–6. doi: 10.1210/jcem.78.2.8106606
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Witchel SF, Azziz R. Nonclassic congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2010) 2010:625105. doi: 10.1186/1687-9856-2010-625105
PubMed Abstract | CrossRef Full Text | Google Scholar
9. KC, Cheng KC, ny MI. Steroid 21-hydroksylasemangel: genotype kan ikke forudsige fænotype. J Clin Endocrinol Metab. (1995) 80:2322–9. doi: 10.1210 / jc.80.8.2322
CrossRef fuldtekst / Google Scholar
10. Hvid PC. Neonatal screening for medfødt adrenal hyperplasi. Nat Rev Endocrinol. (2009) 5:490–8. doi: 10.1038 / nrendo.2009.148
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Kohn B, Levine LS, Pollack MS, Pang S, Lorenzen F, Levy D, et al. Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. (1982) 55:817–27. doi: 10.1210/jcem-55-5-817
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Moran C, Carmina E, Carmina E, Carmina e, Frussetti F, iba Christ L, et al. 21-Ikke-klassisk adrenal hyperplasi er en progressiv lidelse: en multicenterundersøgelse. Er J Fødselslæge Gynecol. (2000) 183:1468–74. doi: 10.1067 / mob.2000.108020
PubMed abstrakt | CrossRef fuldtekst | Google Scholar
13. Jeg er en af dem, der er en af dem, der er en af dem. Screening for 21-hydroksylase-mangelfuld ikke-klassisk binyrehyperplasi blandt hyperandrogene kvinder: en prospektiv undersøgelse. Fertil Steril. (1999) 72:915–25. doi: 10.1016/S0015-0282(99)00383-0
PubMed abstrakt | CrossRef Fuld tekst | Google Scholar
14. Livadas S, Dracopoulou M, Dastamani A, Sertedaki A, Maniati-Christidi M, Magiakou A-M, et al. Spektret af kliniske, hormonelle og molekylære fund hos 280 individer med ikke-klassisk medfødt binyrehyperplasi forårsaget af mutationer af CYP21A2-genet. Clin Endocrinol (Oksf). (2015) 82:543–9. doi: 10.1111 / cen.12543
PubMed abstrakt | CrossRef fuldtekst | Google Scholar
15. Bidet M, Bellann Chantelot C, Galand-Portier M-B, Tardy V, Billaud L, Laborde K, et al. Klinisk og molekylær karakterisering af en kohorte på 161 ikke-relaterede kvinder med ikke-klassisk medfødt binyrehyperplasi på grund af 21-hydroksylasemangel og 330 familiemedlemmer. J Clin Endocrinol Metab. (2009) 94:1570–8. doi: 10.1210 / jc.2008-1582
PubMed abstrakt | CrossRef fuldtekst | Google Scholar
16. Escobar-Morreale HF, Sanch Larsen R, San Mill Larsen JL. En prospektiv undersøgelse af forekomsten af ikke-klassisk medfødt binyrehyperplasi blandt kvinder med hyperandrogen symptomer og tegn. J Clin Endocrinol Metab. (2008) 93:527–33. doi: 10.1210 / jc.2007-2053
PubMed abstrakt | CrossRef fuldtekst | Google Scholar
17. D, Vanighem-Haudiet MC, Sainsard C, Buvat J, Cappoen JP, Ardaens K, et al. Kliniske og biologiske fænotyper ved sen debut af 21-hydroksylasemangel. J Clin Endocrinol Metab. (1986) 63:418–23. doi: 10.1210 / jcem-63-2-418
PubMed abstrakt | CrossRef fuldtekst | Google Scholar
18. (2011). Er basal serum 17-OH progesteron en pålidelig parameter til at forudsige ikke-klassisk medfødt adrenal hyperplasi i for tidlig adrenarche? Turk J Pediatr. 53:274–80.
PubMed abstrakt / Google Scholar
19. F. eks.er der tale om et af de mest almindelige spørgsmål, der er blevet rejst. Medfødt adrenal hyperplasi på grund af steroid 21-hydroksylasemangel: en retningslinje for klinisk praksis i det endokrine samfund. J Clin Endocrinol Metab. (2010) 95:4133–60. doi: 10.1210 / jc.2009-2631
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Dacou-Voutetakis C, Dracopoulou M. High incidence of molecular defects of the CYP21 gene in patients with premature adrenarche. J Clin Endocrinol Metab. (1999) 84:1570–4. doi: 10.1210/jc.84.5.1570
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Det er en af de mest almindelige årsager til, at en person er en person, der ikke er i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at leve. Diagnosen af ikke-klassisk medfødt hyperplasi i binyrebarken på grund af 21-hydroksylasemangel, baseret på serumbasal eller post-ACTH-stimulering 17-hydroksiprogesteron, kan føre til falsk-positiv diagnose. Clin Endocrinol (Oksf). (2016) 84:23–9. doi: 10.1111 / pris 12935
PubMed abstrakt | CrossRef fuldtekst | Google Scholar
22. Det er en af de mest almindelige årsager til, at en person er en person, der ikke er i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at være i stand til at leve. LC-MS / MS forbedrer screeningen mod 21-hydroksylasmangel. Gynecol Endocrinol. (2015) 31:296–300. doi: 10.3109/09513590.2014.994599
PubMed abstrakt | CrossRef Fuld tekst | Google Scholar
23. bbc, CC, Goldman MM, Jang K, Clark-N, tilbage til CK. Samtidig måling af tretten steroidhormoner hos kvinder med polycystisk ovariesyndrom og kontrol kvinder ved hjælp af væskekromatografi-tandem massespektrometri. PLoS One. (2014) 9:e93805. doi: 10.1371 / tidsskrift.pone.0093805
PubMed abstrakt | CrossRef fuldtekst/Google Scholar
24. Ydelsesforbedring ved måling af 5 endogene steroider ved LC-MS/MS kombineret med differentiel ionmobilitetsspektrometri. Clin Chim Acta. (2015) 438:330–6. doi: 10.1016 / j. cca.2014.07.036
PubMed abstrakt | CrossRef Fuld tekst | Google Scholar
25. Stoupa A, P. P., Samara-Boustani D, Thalassinos C, Flechtner i, et al. Utilstrækkelig cortisolrespons på tetracosactid (Synacthen Larus) test I ikke-klassisk medfødt adrenal hyperplasi: en undtagelse fra reglen? Horm Res Paediatr. (2015) 83:262–7. doi: 10.1159/000369901
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. (1992) 90:584–95. doi: 10.1172/JCI115897
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Temeck JV, Pang SY, Nelson C, ny MI. Genetiske defekter af steroidogenese i for tidlig pubarche. J Clin Endocrinol Metab. (1987) 64:609–17. doi: 10.1210 / jcem-64-3-609
PubMed abstrakt | CrossRef fuldtekst | Google Scholar
28. Voutilainen R, J Kurtskel Kurtinen J. For tidlig adrenarche: etiologi, kliniske fund og konsekvenser. J Steroid Biochem Mol Biol. (2015) 145:226–36. doi: 10.1016 / j. jsbmb.2014.06.004
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Oberfield SE, Sopher AB, Gerken AT. Approach to the girl with early onset of pubic hair. J Clin Endocrinol Metab. (2011) 96:1610–22. doi: 10.1210/jc.2011-0225
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Janus D, Wojcik M, Malunowicz E, Starzyk JB. Et tilfælde af tilbagevendende labial adhæsioner i et 15 måneder gammelt barn med asymptomatisk ikke-klassisk medfødt binyrehyperplasi på grund af 21-hydroksylasemangel. J Pediatr Endocrinol Metab. (2012) 25:1017–21. doi: 10.1515 / jpem-2012-0190
PubMed abstrakt | CrossRef Fuld tekst | Google Scholar
31. Gopal-Kothandapani JS, Petkar A, O ‘ Shea E, Banerjee I. perianalt hår som en usædvanlig præsentation af ikke-klassisk medfødt adrenal hyperplasi. BMJ sag Rep. (2013) 2013: bcr2013010123. doi: 10.1136/bcr-2013-010123
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Falhammar H, Nordenström A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine. (2015) 50:32–50. doi: 10.1007/s12020-015-0656-0
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Chrousos GP, Loriaux DL, Mann DL, Cutler GB. Late-onset 21-hydroxylase deficiency mimicking idiopathic hirsutism or polycystic ovarian disease. Ann Intern Med. (1982) 96:143–8. doi: 10.7326/0003-4819-96-2-143
CrossRef Full Text | Google Scholar
34. Witchel SF. Non-classic congenital adrenal hyperplasia. Steroids. (2013) 78:747–50. doi: 10.1016/j.steroids.2013.04.010
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Carmina E, Rosato F, Jannì A, Rizzo M, Longo RA. Extensive clinical experience: relative prevalence of different androgen excess disorders in 950 women referred because of clinical hyperandrogenism. J Clin Endocrinol Metab. (2006) 91:2–6. doi: 10.1210/jc.2005-1457
PubMed Abstract | CrossRef Full Text | Google Scholar
36. H. C., H. C., H. S. S., Rohmer V., T. D., et al. Reproduktivt resultat af kvinder med 21-hydroksylase-mangelfuld ikke-klassisk adrenal hyperplasi. J Clin Endocrinol Metab. (2006) 91:3451–6. doi: 10.1210 / jc.2006-0062
PubMed abstrakt | CrossRef fuldtekst | Google Scholar
37. Levin JH, Carmina E, Lobo RA. Er den uhensigtsmæssige gonadotropinsekretion hos patienter med polycystisk ovariesyndrom svarende til den hos patienter med medfødt adrenal hyperplasi hos voksne? Fertil Steril. (1991) 56:635–40. doi: 10.1016/S0015-0282(16)54592-0
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Carmina E, Lobo RA. Ovarian suppression reduces clinical and endocrine expression of late-onset congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Fertil Steril. (1994) 62:738–43. doi: 10.1016/S0015-0282(16)56998-2
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Ludvig E. klassificering af de typer androgen alopeci (almindelig skaldethed), der forekommer hos det kvindelige køn. Br J Dermatol. (1977) 97:247–54. doi: 10.1111 / j. 1365-2133.1977.tb15179.
PubMed abstrakt | CrossRef fuldtekst/Google Scholar
40. Trakakis E, Papadavid E, Dalamaga M, Koumaki D, Stavrianeas N, Rigopoulos D, et al. Prævalens af ikke-klassisk medfødt adrenal hyperplasi på grund af 21-hydroksylasemangel hos græske kvinder med acne: en hospitalsbaseret tværsnitsundersøgelse. J Eur Acad Dermatol Venereol. (2013) 27:1448–51. doi: 10.1111 / j. 1468-3083.2012. 04613.
PubMed abstrakt | CrossRef fuldtekst/Google Scholar
41. Arlt med, vil DS, vilde SH, Krone N, Doherty EJ, Hahner S, et al. Sundhedsstatus for voksne med medfødt binyrehyperplasi: en kohortestudie af 203 patienter. J Clin Endocrinol Metab. (2010) 95:5110–21. doi: 10.1210 / jc.2010-0917
PubMed abstrakt | CrossRef fuldtekst | Google Scholar
42. Hyperinsulinæmi og insulinfølsomhed hos kvinder med ikke-klassisk medfødt adrenal hyperplasi på grund af 21-hydroksylasemangel: forholdet mellem serumleptinniveauer og kronisk hyperinsulinæmi. Horm Res. (2005) 63:270-4. doi: 10.1159/000086363
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Speiser PW, Serrat J, New MI, Gertner JM. Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1992) 75:1421–4. doi: 10.1210/jc.75.6.1421
PubMed Abstract | CrossRef Full Text | Google Scholar
44. Han TS, Stimson RH, Rees DA, Krone N, Vilis DS, Conge GS, et al. Glukokortikoidbehandling og helbredsresultater hos voksne med medfødt adrenal hyperplasi. Clin Endocrinol. (2013) 78:197–203. doi: 10.1111 / cen.12045
PubMed abstrakt | CrossRef fuldtekst | Google Scholar
45. Van Staa TP, Leufkens HG, Abenhaim L, Jang B, Cooper C. Anvendelse af orale kortikosteroider og risiko for brud. J Bone Miner Res. (2000) 15: 993-1000. doi: 10.1359 / jbmr.2000.15.6.993
PubMed Abstract | CrossRef Full Text | Google Scholar
46. Nebesio TD, Renbarger JL, Nabhan ZM, Ross SE, Slaven JE, Li L, et al. Differential effects of hydrocortisone, prednisone, and dexamethasone on hormonal and pharmacokinetic profiles: a pilot study in children with congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2016) 2016:17. doi: 10.1186 / s13633-016-0035-5
PubMed abstrakt | CrossRef fuldtekst | Google Scholar
47. Ikke-klassisk 21-hydroksylasemangel i barndom og barndom: effekten af tidspunktet for påbegyndelse af terapi på puberteten og den endelige højde. EUR J Endocrinol. (1997) 136:188–95. doi: 10.1530 / eje.0.1360188
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Bonfig W, Bechtold S, Schmidt H, Knorr D, Schwarz HP. Reduced final height outcome in congenital adrenal hyperplasia under prednisone treatment: deceleration of growth velocity during puberty. J Clin Endocrinol Metab. (2007) 92:1635–9. doi: 10.1210/jc.2006-2109
PubMed Abstract | CrossRef Full Text | Google Scholar
49. Dörr HG. Vækst hos patienter med klassisk medfødt adrenal hyperplasi på grund af 21-hydroksylasemangel. Horm Res. (2007) 68(Suppl 5): 93-9. doi: 10.1159 / 000110587
PubMed abstrakt | CrossRef Fuld tekst | Google Scholar
50. Patienter med klassisk medfødt adrenal hyperplasi på grund af 21-hydroksylasemangel kan nå deres målhøjde: Leipsig-oplevelsen. Horm Res. (2008) 70:42-50. doi: 10.1159/000129677
PubMed Abstract | CrossRef Full Text | Google Scholar
51. van der Kaay D, van den Akker E. Ultralow-dose dexamethasone to preserve endogenous cortisol stress response in nonclassical congenital adrenal hyperplasia: a new promising treatment. Int J Endocrinol Metab. (2014) 12:e14657. doi: 10.5812/ijem.14657
CrossRef Full Text | Google Scholar
52. Lin-Su K, Vogiatzi MG, Marshall I, Harbison MD, Macapagal MC, Betensky B, et al. Treatment with growth hormone and luteinizing hormone releasing hormone analog improves final adult height in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. (2005) 90:3318–25. doi: 10.1210/jc.2004-2128
PubMed Abstract | CrossRef Full Text | Google Scholar
53. McCann-Crosby B, Chen M-J, Lyons SK, Lin Y, Axelrad M, Dietrich JE, et al. Nonclassical congenital adrenal hyperplasia: mål for behandling og overgang. Pediatr Endocrinol Rev. (2014) 12: 224-38.
PubMed abstrakt | Google Scholar
54. Ogilvie CM, Crouch NS, Rumsby G, Creighton SM, Liao L-M, Conge GS. Medfødt adrenal hyperplasi hos voksne: en gennemgang af medicinske, kirurgiske og psykologiske problemer. Clin Endocrinol. (2006) 64:2–11. doi: 10.1111 / j. 1365-2265.2005.02410.x
PubMed Abstract | CrossRef Full Text | Google Scholar
55. Mah PM, Jenkins RC, Rostami-Hodjegan A, Newell-Price J, Doane A, Ibbotson V, et al. Weight-related dosing, timing and monitoring hydrocortisone replacement therapy in patients with adrenal insufficiency. Clin Endocrinol. (2004) 61:367–75. doi: 10.1111/j.1365-2265.2004.02106.x
PubMed Abstract | CrossRef Full Text | Google Scholar
56. Breech LLAA af PC på en, pleje AC af O og GC på AH. Menstruation hos piger og unge: brug af menstruationscyklussen som et vigtigt tegn. Pædiatri. (2006) 118:2245–50. doi: 10.1542 / peds.2006-2481
CrossRef fuldtekst | Google Scholar
57. Merke DP, Poppas DP. Behandling af unge med medfødt adrenal hyperplasi. lancet Diabetes Endocrinol. (2013) 1:341–52. doi: 10.1016/S2213-8587(13)70138-4
PubMed Abstract | CrossRef Full Text | Google Scholar
58. Verma S, Green-Golan L, VanRyzin C, Drinkard B, Mehta SP, Weise M, et al. Adrenomedullary function in patients with nonclassic congenital adrenal hyperplasia. Horm Metab Res. (2010) 42:607–12. doi: 10.1055/s-0030-1253385
PubMed Abstract | CrossRef Full Text | Google Scholar
59. New MI. Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2006) 91:4205–14. doi: 10.1210/jc.2006-1645
PubMed Abstract | CrossRef Full Text | Google Scholar
60. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2018) 103:4043–88. doi: 10.1210/jc.2018-01865
PubMed Abstract | CrossRef Full Text | Google Scholar
61. Kruse B, Riepe FG, Krone N, Bosinski HAG, Kloehn S, Partsch CJ, et al. Congenital adrenal hyperplasia – how to improve the transition from adolescence to adult life. Exp Clin Endocrinol Diabetes. (2004) 112:343–55. doi: 10.1055/s-2004-821013
PubMed Abstract | CrossRef Full Text | Google Scholar
62. Gastaud F, Bouvattier C, Duranteau L, Brauner R, Thibaud E, Kutten F, et al. Forringede seksuelle og reproduktive resultater hos kvinder med klassiske former for medfødt adrenal hyperplasi. J Clin Endocrinol Metab. (2007) 92:1391–6. doi: 10.1210 / jc.2006-1757
PubMed abstrakt | CrossRef fuldtekst | Google Scholar
63. M, Muthusamy K, Smushkin G, LAMPROPULOS JF, ELAMIN MB, Abu Elnour NO, et al. Anvendelse til forebyggelse af virilisering i graviditeter med risiko for klassisk medfødt adrenal hyperplasi på grund af 21-hydroksylase (CYP21A2) mangel: en systematisk gennemgang og metaanalyser. Clin Endocrinol. (2010) 73:436–44. doi: 10.1111 / j. 1365-2265.2010. 03826.
CrossRef Fuld tekst / Google Scholar
64. Bidet M, Bellann Chantelot C, Galand-Portier MB, Golmard JL, Tardy V, Morel Y, et al. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2010) 95:1182–90. doi: 10.1210/jc.2009-1383
PubMed Abstract | CrossRef Full Text | Google Scholar
65. Birnbaum MD, Rose LI. Late onset adrenocortical hydroxylase deficiencies associated with menstrual dysfunction. Obstet Gynecol. (1984) 63:445–51.
PubMed Abstract | Google Scholar
66. Cuhaci N, Aydin C, Yesilyurt A, Pinarli FA, Ersoy R, Cakir B. Nonclassical congenital adrenal hyperplasia and pregnancy. Case Rep Endocrinol. (2015) 2015:296924. doi: 10.1155/2015/296924
PubMed Abstract | CrossRef Full Text | Google Scholar
67. Deneux C, Tardy V, Anne D, Mornet E, Billaud L, Charron D, et al. Fænotype-genotype korrelation hos 56 kvinder med ikke-klassisk medfødt adrenal hyperplasi på grund af 21-hydroksylasemangel. J Clin Endocrinol Metab. (2001) 86:207–13. doi: 10.1210 / jcem.86.1.7131
PubMed abstrakt | CrossRef fuldtekst | Google Scholar
68. Simonetti L, bruk CD, bregne af CS, Benavides-Mori B, Delea M, Kolomenski JE, et al. CYP21A2-mutationsopdatering: omfattende analyse af databaser og offentliggjorte genetiske varianter. Hum Mutat. (2018) 39:5–22. doi: 10.1002 / humu.23351
PubMed Abstract | CrossRef Full Text | Google Scholar
69. OMIM Entry – * 613815 – CYTOCHROME P450 FAMILY 21 SUBFAMILY A POLYPEPTIDE 2; CYP21A2.
Google Scholar
70. Miller WL, Witchel SF. Prenatal treatment of congenital adrenal hyperplasia: risks outweigh benefits. Am J Obstet Gynecol. (2013) 208:354–9. doi: 10.1016/j.ajog.2012.10.885
PubMed Abstract | CrossRef Full Text | Google Scholar
71. Lekarev O, New MI. Adrenal disease in pregnancy. Best Pract Res Clin Endocrinol Metab. (2011) 25:959–73. doi: 10.1016/j.beem.2011.08.004
PubMed Abstract | CrossRef Full Text | Google Scholar
72. New MI, Abraham M, Yuen T, Lekarev O. An update on prenatal diagnosis and treatment of congenital adrenal hyperplasia. Semin Reprod Med. (2012) 30:396–9. doi: 10.1055/s-0032-1324723
PubMed Abstract | CrossRef Full Text | Google Scholar
73. Kazmi D, Bailey J, Yau M, Abu-Amer W, Kumar A, Low M, et al. New developments in prenatal diagnosis of congenital adrenal hyperplasia. J Steroid Biochem Mol Biol. (2017) 165:121–3. doi: 10.1016/j.jsbmb.2016.06.016
PubMed Abstract | CrossRef Full Text | Google Scholar
74. Clayton PE, Miller VL, Oberfield SE, Sippell, Speiser PV, et al. Konsensus Erklæring om 21-hydroksylasemangel fra Det Europæiske Selskab for pædiatrisk endokrinologi. Horm Res. (2002) 58:188-95. doi: 10.1159 / 000065490
PubMed abstrakt | CrossRef Fuld tekst | Google Scholar
75. Nandagopal R, Sinaii N, Avila NA, Van Rysin C, Chen M, Finkielstain GP, et al. Fænotypisk profilering af forældre med kryptisk ikke-klassisk medfødt adrenal hyperplasi: fund i 145 ikke-relaterede familier. EUR J Endocrinol. (2011) 164:977–84. doi: 10.1530 / EJE-11-0019
PubMed abstrakt | CrossRef Fuld tekst | Google Scholar
76. Takasu N, Nakachi K, Higa H. udvikling af Graves’ hyperthyroidisme forårsagede en binyrekrise hos en patient med tidligere ukendt ikke-klassisk 21-hydroksylasemangel. Praktikant Med. (2010) 49:1395–400. doi: 10.2169 / internalmedicin.49.3573
PubMed Abstract | CrossRef Full Text | Google Scholar
77. Meyer-Bahlburg HFL, Dolezal C, Baker SW, Carlson AD, Obeid JS, New MI. Prenatal androgenization affects gender-related behavior but not gender identity in 5-12-year-old girls with congenital adrenal hyperplasia. Arch Sex Behav. (2004) 33:97–104. doi: 10.1023/B:ASEB.0000014324.25718.51
CrossRef Full Text | Google Scholar
78. Meyer-Bahlburg HFL, Dolesal C, bager SV, ny MI. Seksuel orientering hos kvinder med klassisk eller ikke-klassisk medfødt adrenal hyperplasi som en funktion af graden af prænatal androgenoverskud. Arch Køn Opføre Sig. (2008) 37:85–99. doi: 10.1007 / s10508-007-9265-1
PubMed abstrakt / CrossRef Fuld tekst / Google Scholar