sclera albastră este cauzată de subțierea și transparența fibrelor de colagen ale sclerei care permit vizualizarea uvea subiacente (Figura 1). Sclera este stratul exterior alb al ochiului, care înconjoară irisul poate fi subțiat în boli congenitale, cum ar fi osteogeneza imperfectă 1). Alte boli asociate cu sclera albastră includ tulburări multiple ale țesutului conjunctiv, cum ar fi sindromul corneei fragile 2), sindromul Marshall și Stickler 3), poezii (polineuropatie, organomegalie, endocrinopatie, proteine monoclonale și modificări ale pielii) sindromul 4), sindromul Marfan, sindromul Ehlers–Danlos, pseudoxanthoma elasticum și sindromul Willems de Vries, pentru a numi câteva. Tulburările osoase și ale sângelui includ, de asemenea, anemia Diamond–Blackfan, anemia severă cu deficit de fier, boala Paget juvenilă și deficitul de fosfatază acidă 5).
formele Severe de osteogeneză imperfectă sunt cel mai adesea diagnosticate la începutul vieții, dar cazurile ușoare nu pot fi observate decât mai târziu în viață. Culoarea albastru-gri a sclerei se datorează venelor coroidiene subiacente care se manifestă. Acest lucru se datorează faptului că sclera este mai subțire decât în mod normal, deoarece colagenul defect de tip I nu se formează corect 6). Sclera este o structură densă de țesut conjunctiv slab vascularizată compusă din tipurile I, III, IV, V, VI și VIII colagen, precum și elastină, proteoglicani și glicoproteine 7).în Statele Unite, incidența osteogenezei imperfecte este estimată a fi una la 20.000 de nașteri vii. Se estimează că 20.000-50.000 de persoane sunt afectate de osteogeneza imperfectă în Statele Unite 8).
persoanele cu osteogeneză imperfectă se nasc cu țesut conjunctiv defect sau fără capacitatea de a-l face, de obicei din cauza unei deficiențe de colagen de tip I. Această deficiență apare dintr-o substituție de aminoacizi a glicinei cu aminoacizi mai voluminoși în structura triplă helix a colagenului. Ca urmare, organismul poate răspunde prin hidrolizarea structurii necorespunzătoare a colagenului 9). Dacă organismul nu distruge colagenul necorespunzător; relația dintre fibrilele de colagen și cristalele de hidroxiapatită pentru a forma osul este modificată, provocând fragilitate. Ca tulburare genetică, osteogeneza imperfectă a fost privită istoric ca o tulburare dominantă autosomală a colagenului de tip I. Majoritatea cazurilor au fost cauzate de mutații ale genelor COL1A1 și COL1A2 10). În ultimii ani, a existat identificarea formelor autosomale recesive. Au fost descrise cel puțin șapte subseturi, deși patru subtipuri majore sunt cele mai frecvente și variază de la ușoare la severe. Persoanele cu osteogeneză imperfectă de tip I au o deformare osoasă mică, sclera albastră persistentă, aproape de înălțimea normală până la vârsta adultă și o >50% șanse de pierdere a auzului până la vârsta adultă. Pacienții cu osteogeneză imperfectă letală perinatală (tip II) prezintă cea mai mare severitate, cu fracturi multiple in utero sau de la naștere. Acești pacienți sunt de obicei născuți morți sau mor devreme. Severitatea osteogenezei imperfecte depinde de defectul genei specifice. Majoritatea cazurilor de osteogeneză imperfectă sunt moștenite de la un părinte. Cu toate acestea, unele cazuri sunt rezultatul unor noi mutații genetice. O persoană cu osteogeneză imperfectă are o șansă de 50% de a transmite Gena și boala copiilor săi 11).
Figura 1. Anatomia ochilor
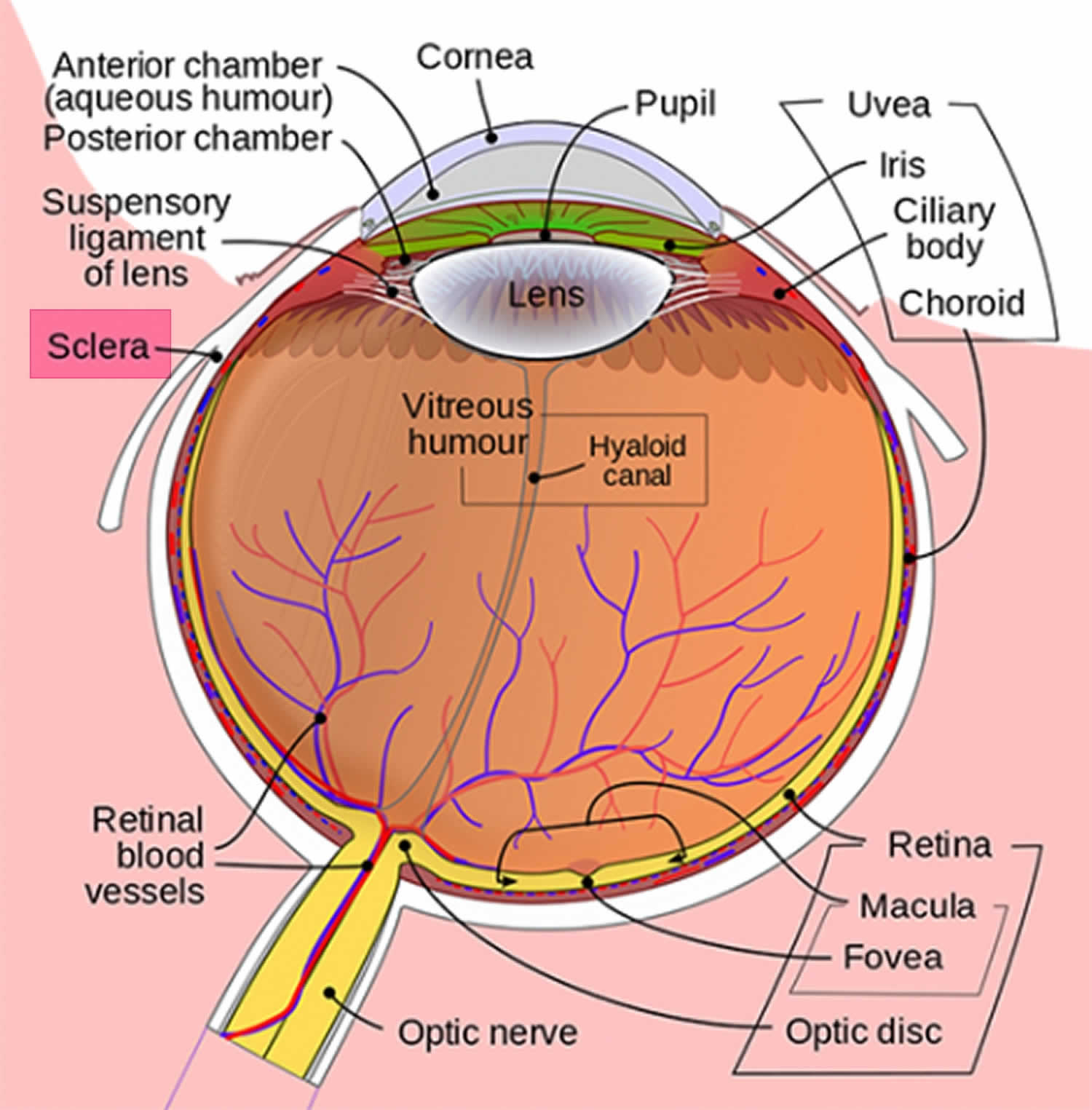
Figura 2. Sclera albastră
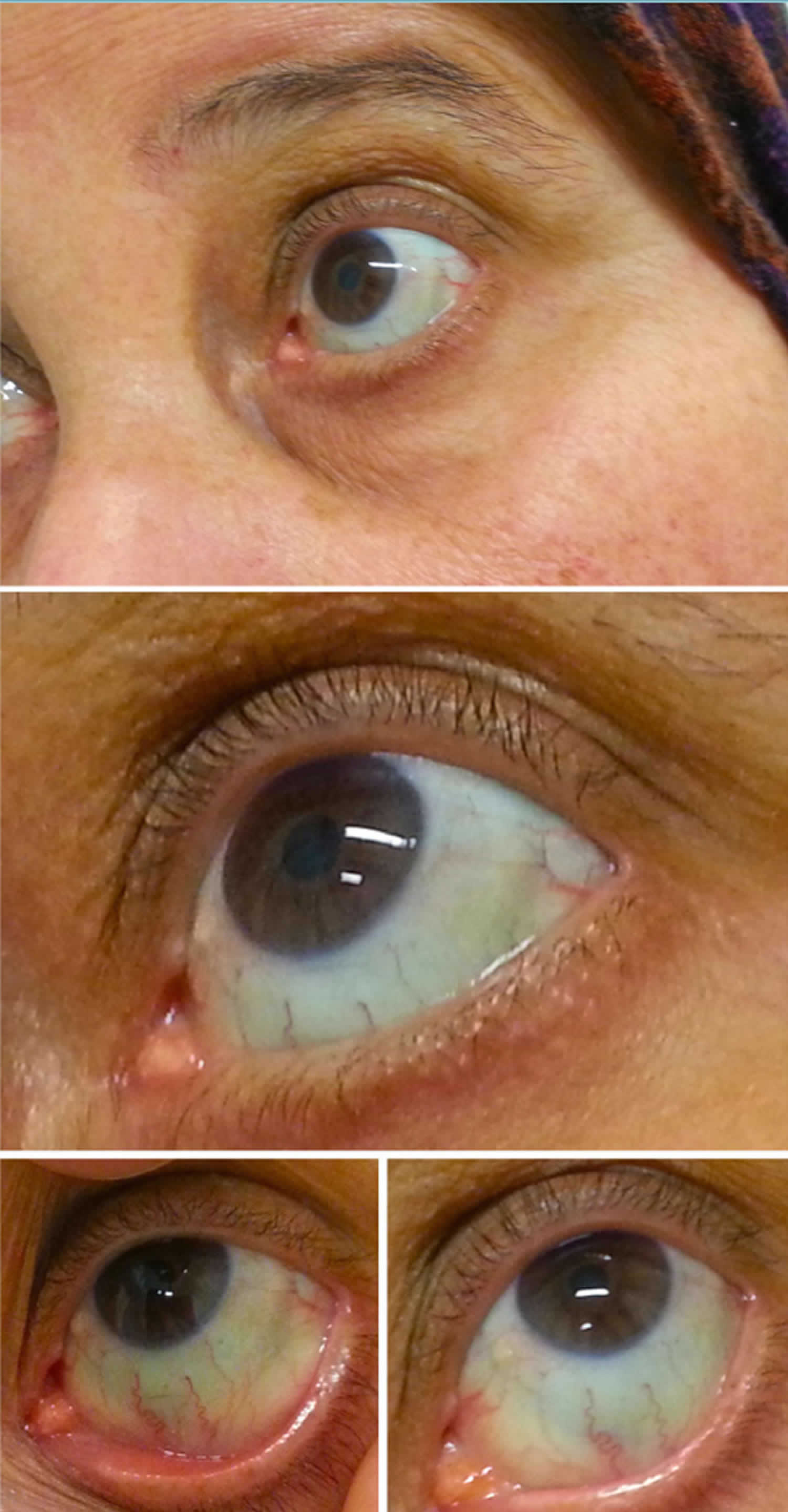
sclera albastră provoacă
sclera albastră este cea mai consistentă manifestare a osteogenezei imperfecte, care rezultă dintr-o mutație în COLIA1 și COL1A2, codificând procolagenul de tip I. Cu toate acestea, clasificările acestei afecțiuni (tipurile IV–VI) au fost identificate cu sclere normale. Osteogeneza imperfectă este, de asemenea, asociată cu fragilitatea anormală a oaselor și surditatea.
corneea fragilă, sclera albastră și părul roșu sunt asociate cu sindromul corneei fragile, o afecțiune care prezintă, de asemenea, anomalii scheletice,dentare și ale pielii 13). S-a constatat că o mutație missense în ZNF469 este cauzală pentru boală.
alte afecțiuni subiacente asociate cu formarea sclerei albastre includ sindromul Ehlers-Danlos (tip VI), pseudoxanthoma elasticum, plana corneei, sclerocornea periferică, buphthalmos, keratoconus, keratoglobus, miopie ridicată, stafilom ciliar / ecuatorial, melanocitoză oculodermică și microcornea. Rar, sclera albastră apare cu sindromul Hallermann-Streiff, sindromul Marfan, sindromul Turner, sindromul Cheney, sindromul Menkes, pyknodysostosis, cornee fragilă sau displazie ectodermică.
sclera albastră poate apărea, de asemenea, la sugarii normali în primele câteva luni de viață; cu toate acestea, persistența decolorării albastre în timp poate sugera prezența presiunii intraoculare crescute. Sugarii prematuri prezintă frecvent sclere albastre, în special cele de origine caucaziană.
sclera albastră poate apărea, de asemenea, izolat ca o anomalie autosomală dominantă sau autosomală recesivă moștenită 14).
simptome sclera albastră
sclera albastră prezintă un aspect albăstrui la sclera și poate fi asociată cu cauză patologică sau non-patologică. Alte caracteristici oculare ale tulburărilor țesutului conjunctiv asociate cu sclera albastră includ corneea subțire, pliul epicantal, miopia, keratoconusul și dungile angioide. Caracteristicile sistemice ale tulburărilor țesutului conjunctiv asociate cu sclera albastră includ anomalii ale pielii, anomalii cardiace, cifoscolioză, hipermobilitate articulară, oase fragile, anomalii ale auzului, anomalii vasculare și anomalii gastro-intestinale 15).
diagnosticul sclerei albastre
evaluarea diagnosticului sclerei albastre implicăexaminarea externă, biomicroscopia lămpii cu fantă și evaluarea sistemică a tulburărilor asociate.Deși nu există un test definitiv pentru osteogeneza imperfectă, testarea genetică poate confirma sau exclude mutațiile cunoscute 16).
tratamentul sclerei albastre
tratamentul pentru sclera albastră implică diagnosticarea și tratarea cauzei care stă la baza acesteia. Sclera albastră se datorează în principal sindroamelor genetice și, într-o măsură mai mică, tulburărilor nongenetice și poate apărea ca efect secundar al consumului de medicamente. Sclerele albastre sunt frecvent asociate cu tulburări congenitale de sinteză a colagenului, cum ar fi osteogeneza imperfectă, sindromul Marfan, sindromul Ehlers–Danlos, pseudoxanthoma elasticum și sindromul Willems de Vries, pentru a numi câteva. Tulburările osoase și ale sângelui includ, de asemenea, anemia Diamond–Blackfan, anemia severă cu deficit de fier, boala Paget juvenilă și deficitul de fosfatază acidă 17). Sclerele albastre dobândite au fost descrise la adulți cu melanoză oculară, melanom conjunctival, alcaptonurie și boală Addison. Pot fi indicate trimiteri adecvate la evaluări pediatrice și ortopedice pentru manifestări non-oculare 18).
consilierea genetică pentru bolile moștenite asociate poate fi benefică pentru pacienții cu scleroză albastră și alte manifestări ale bolii sistemice.
intervenția chirurgicală poate fi indicată în cazurile de subțiere și perforare extremă. Suportul Structural poate fi asigurat de o grefă sclerală conservată saufascia utologă lata, în special în cazurile care necesită suturarea unui dispozitiv, cum ar fi un implant tubular 19). Investigațiile sistemice și tratamentul trebuie luate în considerare pentru a aborda patologia de bază.
prognosticul sclerei albastre
prognosticul variază în funcție de prezența manifestărilor oculare și sistemice ale tulburării de bază. Pacienții cu sclera albastră prezintă un risc crescut de ruptură a globului sau perforație sclerală intraoperatorie în timpul intervenției chirurgicale de rutină a ochilor. Tulburările sistemice sunt, de asemenea, predominante la pacienți,cum ar fi fistula carotidă-cavernoasă, ruptura arterială, pierderea auzului și fracturile osoase 20).
referințe