- Definições e Prevalência
- o Diagnóstico
- manifestações na adolescência e na vida adulta
- NCCAH e Reprodução
- Infertilidade
- tratamento pré-natal de CAH
- outras questões especiais
- controlo do Stress na NCCAH
- aspectos Psicobiológicos na NCCAH
- conclusões
- contribuições do autor
- Declaração de conflito de interesses
Definições e Prevalência
a hiperplasia adrenal Congênita (CAH) engloba uma família de herança autossômica recessiva transtornos caracterizados por leve a acentuada deficiência de cortisol síntese, devido a uma deficiência em uma das cinco adrenal steroidogenic enzimas necessárias para a produção de cortisol (1, 2). Convencionalmente, a CAH é dividido em (a) clássica (CCAH), apresentando-se com sal, desperdício ou a simples virilizantes forma que se manifesta no nascimento e/ou no período neonatal, e (b) não-clássica (NCCAH), representando uma forma menos grave da doença que carece de ambigüidade genital, não é imediatamente com risco de vida, e apresenta-se mais tarde na vida, ou permanece assintomática, ou seja, é diagnosticada como uma doença distinta (3). No entanto, as fronteiras entre as diferentes formas de CAH não são estritamente definidas, o que tende a aumentar os desafios associados a esta desordem. É, portanto, aconselhável considerar a HCA como um continuum de fenotipos, de severos a ligeiros ou assintomáticos (3).a forma mais comum de CAH é causada por deficiência de 21-hidroxilase (21OHD), resultando em incapacidade ou não Conversão de 17 hidroxiprogesterona (17 OHP) para 11 deoxicortisol e de progesterona para desoxicorticosterona. O bloqueio da conversão de esteróides resulta no aumento da produção de precursores androgênicos, sob estimulação CRH-ACTH, levando ao hiperandrogenismo bioquímico, marcado por níveis elevados de 17-OHP. A forma clássica da doença, ocorre em 1 de 16.000 nascimentos vivos em todo o mundo. No entanto, deve referir-se que a percepção da incidência da doença está relacionada com o método de rastreio utilizado. Por exemplo, tem sido relatado que a incidência da forma clássica de CAH na Suécia é de 1:11.500 quando uma abordagem de pesquisa de casos é usada, que figura no entanto cai para 1:9.800 quando o rastreio hormonal é aplicado. Da mesma forma, a incidência na França e na Suíça varia entre 1:15,472 e 1:23,000 e 1:10,749 e 1:11,661 quando os diferentes métodos de triagem são usados (4).
NCCAH é muito mais freqüente, ocorrendo em aproximadamente 1 em cada 1.000 Caucasianos e, mais comumente, em certos grupos étnicos, como os Judeus Ashkenazi (1:27), Hispânicos (1:53), Iugoslavos (1:62), e os Italianos (1:300) (5, 6). Não obstante, faltam dados sobre a prevalência da NCCAH com base em diferentes métodos de estimativa (inquérito de casos, rastreio hormonal e análise molecular). A NCCAH é considerada a doença endócrina recessiva autossómica mais comum com uma frequência portadora de 1:25 a 1:10. Na NCCAH devido a 21-OHD, estima-se que a atividade enzimática residual seja de cerca de 10-70% (7-9).
o Diagnóstico
Em comparação com o diagnóstico da forma clássica da doença, o que é feito ao nascimento ou no período neonatal devido a ambigüidade genital e/ou sal de perda de sintomas ou através de programas de rastreio empregados em alguns países a maioria dos casos de NCCAH não são facilmente detectáveis (4, 10). Além disso, muitos indivíduos permanecem assintomáticos durante a infância e adolescência, têm função reprodutora normal, e só se tornam conscientes da NCCAH devido ao diagnóstico de outro membro da família e conseqüente teste (11). No entanto, a maioria das mulheres com NCCAH procuram assistência médica quando experimentam sintomas de excesso de andrógeno e, quando a suspeita clínica pede testes, níveis elevados de OHP basal 17 provavelmente não apontam para um diagnóstico de NCCA.de facto, as directrizes clínicas propostas pela Sociedade Endócrina recomendam um valor não estimulado de base de 17 OHP como ferramenta de rastreio para a NCCAH. Manhã 17 níveis de OHP >6 nmol/L na fase folicular nas fêmeas menstruadas deve levar a uma avaliação mais aprofundada, uma vez que foi demonstrado que níveis acima de 6 nmol / L capturam 90% dos indivíduos NCAH (12, 13). As medições aleatórias de 17 OHP não têm mostrado ser úteis, uma vez que estes frequentemente produzem níveis normais em pacientes com NCCAH, e eles são, além disso, extremamente elevados na fase lútea em metade das mulheres saudáveis (13). No entanto, em nossos dados derivados de 280 indivíduos com a doença, seis pacientes (2.1%) tinha um valor basal de 17 OHP < 6 nmol / L (14). Além disso, Bidet et al., numa grande coorte de mulheres com NCCAH verificada por técnicas moleculares, também encontraram valores basais de 17 OHP inferiores a 6 nmol / L em 8% dos indivíduos estudados (15). Por último, com base nos dados recolhidos por Speiser et al., 9% dos indivíduos com NCCAH apresentaram valores de 17 OHP inferiores a 2 ng/ml (o que corresponde a 6nmol / L). De acordo com outros estudos, um valor inicial de 17 OHP entre 5, 1 e 9 nmol/L é suficiente para o diagnóstico da NCCAH (13, 16, 17). Recentemente, um nível basal de 17 OHP de 4.6 nmol/L foi sugerido como um limiar para os testes da ACTH para prever a NCCAH em indivíduos com adrenarche prematura durante a infância (18).
a soma total destes resultados e sugestões indica que a selecção dos doentes que serão submetidos a um teste de estimulação Sinactena deve ser avaliada caso a caso. Para o diagnóstico é necessário um nível de 17 OHP pós-estimulação superior a 3 nmol / L (19). Heterozigotos Portadores de uma mutação CYP21A2 apresentam níveis ligeiramente elevados de 17 OHP após estimulação ACTH, embora haja sobreposição em indivíduos não afectados (9). No entanto, como Dacou-Voutetakis et al. ter apontado, se a soma dos valores basais e pós-estimulação 17 OHP exceder 1,5 nmol / L, então a possibilidade de heterozigosidade é excepcionalmente alta (20).outra consideração importante diz respeito às técnicas utilizadas para a avaliação de 17 OHP. 17 os níveis de OHP são medidos por uma variedade de métodos de imunoensaio, mas como foi recentemente mostrado, os resultados mais precisos e confiáveis foram alcançados pela implementação da combinação de cromatografia líquida com espectrometria de massa (LC-MS/MS). Na verdade, foram encontradas muitas medições falsas positivas de 17 OHP quando as medições de LC-MS/Ms foram comparadas com os métodos padrão (21), No entanto, estes últimos procedimentos não são universalmente utilizados, no caso de um diagnóstico incerto, eles fornecerão resultados mais precisos. De notar que os limiares de rastreio e diagnóstico para CL-MS/MS quantificados 17 OHP ainda não estão definidos. É provável que estes limiares sejam inferiores aos estabelecidos com imunoensaios devido à maior especificidade da CL-MS/MS, um método menos propenso à reactividade cruzada e às interferências (22, 23). A questão de saber se é necessário um perfil de esteroides urinários para o diagnóstico definitivo permanece por esclarecer (22). Em casos limítrofes, é aconselhável obter um perfil adrenocortical completo após o teste de estimulação ACTH para diferenciar a deficiência de 21-hidroxilase de outros defeitos enzimáticos e estabelecer um diagnóstico firme.especificamente, deve ser realizado um perfil esteróide completo em casos equívocos para confirmar a deficiência de 21-hidroxilase e excluir outros detectores enzimáticos. A inclusão de 17 OHP, cortisol, 11-deoxycorticosterone,11-deoxycortisol,17-OH-pregnenolona, Dehidroepiandrosterona, e androstenediona em um soro de esteróides perfil seria útil para excluir outras causas de CAH, tais como 11β-hydroxylase de deficiência e, mais raramente, 3ß-hidroxiesteróide enzima deficiência ou P450 oxidoreductase deficiência (24). Embora os testes genéticos não sejam considerados uma ferramenta primária de diagnóstico para a NCCAH neste momento, é obrigatório para confirmação do diagnóstico e para aconselhamento genético (3).no que respeita aos níveis de cortisol, é geralmente esperado um valor pós-estimulação de pelo menos 496 nmol/l. De notar, de acordo com Stoupa et al., 60% das 47 crianças com NCCAH como resultado, de 21 de OHD com baixos valores de cortisol após a estimulação, uma constatação aponta para a necessidade de maior vigilância, para o desenvolvimento de insuficiência adrenal durante grande estressor eventos (25).a expressão clínica da NCCAH é caracterizada por um elevado nível de polimorfismo no que diz respeito não só à idade de início, mas também aos diferentes sinais e sintomas. É relatado que a primeira apresentação clínica da NCCAH é em 11% dos casos antes da idade de 10 anos e em 80% entre as idades de 10 e 40 anos (12). A correlação genótipo-fenótipo na CAH e NCCAH ainda não foi elucidada. Speiser et al. sugerem que a maior parte, mas não toda, da variabilidade fenotípica na deficiência de 21-hidroxilase resulta da variação alélica no CYP21A2 (26).manifestações na infância no período recém-nascido, tipicamente as fêmeas da NCCAH permanecem assintomáticas e têm órgãos genitais externos normais. O primeiro caso de NCCAH relatado é uma menina de 6 meses de idade que desenvolveu pêlos púbicos (11). Geralmente, os achados e sintomas clínicos em casos de NCCAH começam a partir da idade de 5 anos ou mesmo mais tarde e estão relacionados com o aumento dos níveis de androgênio, embora deficiência ligeira cortisol também pode ocorrer em alguns casos (8). Em 92% dos casos de NCCAH, o primeiro sintoma que se manifesta é a pubarche prematura (PP), que ocorre com menos de 8 anos de idade em meninas e com menos de 9 anos em meninos (12), com uma prevalência estimada entre 10 e 11,3% (15). De notar, em crianças referenciadas com PP, de acordo com diferentes estudos, a incidência de NCCAH varia de 5 a 30% (20, 27). No entanto, com base em nossa análise de 280 pacientes com confirmação molecular da NCCAH, descobrimos que a incidência de PP em 94 mulheres com menos de 8 anos foi de 88%. Por outro lado, uma análise de 45 homens com NCCAH identificou PP em apenas 29% dos indivíduos (14). Além disso, devemos ter em mente que cerca de metade dos indivíduos com PP podem ser portadores heterozigotos de uma mutação CYP21A2.outro aspecto que precisa de clarificação é a relação entre PP e puberdade precoce. Em alguns casos, a aromatização periférica de androgénios adrenais a estrogénios pode ativar o eixo hipotalâmico-hipófise-gonadal, levando à puberdade precoce secundária (28). Neste caso, é necessário um acompanhamento regular. Algumas crianças desenvolvem hirsutismo, acne e / ou crescimento rápido e manifestam alta estrutura e avanço da idade óssea (29). Este último pode resultar em altura final truncada como consequência da rápida fusão epifiseal. Outros achados clínicos raros durante a infância incluem aderência labial, cabelo perianal, clitoromegalia e rouquidão de voz (30-32).
manifestações na adolescência e na vida adulta
durante a adolescência e a idade adulta, a NCAH nas mulheres apresenta sintomas hiperandrogénicos semelhantes à síndrome do ovário policístico (PCOS) (33). Os sintomas incluem hirsutismo, acne, alopecia androgênica, anovulação, disfunção menstrual e infertilidade. Num estudo multicêntrico, os sintomas mais comuns entre adolescentes e mulheres adultas foram hirsutismo (59%), oligomenorreia (54%) e acne (33%). Entre 161 mulheres com NCCAH, apresentando sintomas foram hirsutismo (78%), disfunção menstrual (54,7%) e diminuição da fertilidade (12%) (34). No nosso grupo de 161 mulheres com a doença, 63% do paciente apresentou um fenótipo policístico do tipo ovário (14). Este resultado tem uma implicação vice-versa, uma vez que a incidência de NCCAH é elevada em mulheres com hiperandrogenemia. Certamente, o estudo de Witcel et al. relatar uma incidência de NCCAH variando de 1 a 33% em mulheres hiperandrogênicas é de grande interesse. Em outro estudo de 950 mulheres que foram encaminhadas para avaliação do excesso de androgênio, a NCCAH foi detectada em 4, 2% delas. No entanto, os doentes com NCCAH eram mais jovens e hirsutosos em comparação com os outros grupos de doentes hiperandrogénicos. Eles também foram caracterizados por níveis significativamente mais elevados de testosterona, testosterona livre, e 17 OHP do que pacientes com outras síndromes hiperandrogênicas. No ultra-som ovárico, 77% apresentavam ovários poliquísticos e 41% apresentavam um aumento do tamanho do ovário. Em suma, cumpriram os critérios dos OCP, de acordo com a NIH e Roterdão, numa percentagem de 56 e 72,8%, respectivamente (35).
a natureza progressiva da doença é destacada pelo fato de que a prevalência do hirsutismo tem demonstrado aumentar com a idade e tem sido observada ser rara antes da puberdade. O hirsutismo é a manifestação clínica mais comum relatada em pacientes com NCCAH, variando de 71 a 96% (28, 36). Raparigas pubertais com NCCAH tipicamente presentes com hirsutismo (37, 38). No outro extremo do espectro está a questão da alopecia. Em doentes com NCCAH foram notificados casos de descamação do padrão masculino. A prevalência de alopecia também pareceu aumentar com a idade, de 6% em pacientes durante a segunda década de sua vida para 19% no quinto, indicando novamente a natureza progressiva da doença (39). A Acne é notificada em quase 33% dos indivíduos com NCCAH (36). Notavelmente, mutações do gene CYP21A2 causando NCCAH foram detectadas em 4, 9% de 123 mulheres adultas apresentando acne grave (40).as irregularidades menstruais incluindo oligomenorreia ou amenorreia secundária podem muitas vezes ser o sinal de apresentação da NCCAH em indivíduos pós-menarche. Além disso, 8-9% dos casos também experimentam amenorréia primária e, por esta razão, procurar aconselhamento médico pela primeira vez. Além disso, entre os indivíduos NCCAH, oligomenorréia é mais comum do que amenorréia primária. Por exemplo, em uma série de Moran et al. entre as mulheres NCCAH de 10 a 19 anos, a oligomenorreia esteve presente em 56% dos casos, em comparação com apenas 9% da adolescência com amenorreia primária (12). Para concluir, tantos sintomas da NCCAH se assemelham aos dos OCP, que não é surpreendente que a NCCAH tenha sido apelidada de grande mimicker dos OCP. Em qualquer caso, deve ser considerado um diagnóstico de NCCAH durante a avaliação de qualquer mulher jovem que seja encaminhada para sintomas hiperandrogênicos.
em um estudo de Arlt et al., os pacientes com NCCAH foram caracterizados por IMC significativamente maior e menor altura final em comparação com os controles correspondentes (41). Estes dados, em conjunto com alguns estudos que indicam a presença de insensibilidade à insulina, sugerem um perfil metabólico desfavorável que pode ser explicado patofisiologicamente pelo efeito de concentrações androgénicas elevadas e/ou como resultado da terapêutica glucocorticóide (42, 43). A osteoporose também pode ser detectada nestes indivíduos, provavelmente como consequência da terapêutica com corticosteróides (34).uma vez estabelecido o diagnóstico da NCCAH, várias questões relacionadas com as recomendações de tratamento subsequentes merecem consideração. A primeira questão a ser abordada é se a terapia glucocorticóide (GC) está indicada. O raciocínio geral por trás desta abordagem é que ao fornecer concentrações suficientes de cortisol para o paciente, suas necessidades diárias são cobertas e, consequentemente, a estimulação do eixo CRH-ACTH será reduzida, levando à diminuição da produção de andrógenos adrenais. A regra geral relativa a esta abordagem é que os GCs são administrados em doses de substituição e não farmacológica, enquanto a influência da idade, sexo, dados laboratoriais, recomendações específicas do doente e objectivos de tratamento em terapêutica de substituição glucocorticóide são seriamente tomados em consideração. No entanto, também devemos estar cientes de que a terapia de substituição de GC prolongada pode levar a hipercortisolemia, com os efeitos adversos bem documentados resultantes em todos os aspectos do metabolismo, especialmente crescimento, distribuição de gordura, e resistência à insulina, bem como no perfil psicológico. Outra grande desvantagem desta abordagem é a falta de um índice clínico adequado ou marcador bioquímico de dose de substituição adequada, tais como existem em relação aos valores de TSH no hipotiroidismo.por último, o facto de não existir consenso quanto à utilização de GC e a ausência de dados a longo prazo sobre as diferentes modalidades de administração de GC complicam ainda mais as decisões dos médicos que participam na consulta e a selecção da escolha óptima para cada doente. A hidrocortisona é normalmente utilizada em crianças, uma vez que se assemelha mais à hormona natural (cortisol), mas não é considerada uma abordagem adequada em adolescentes e mulheres jovens devido à necessidade de doses múltiplas diárias. Assim, a maioria dos endocrinologistas adultos prefere dexametasona ou prednisolona, em doses apropriadas. Por outro lado, deve salientar-se que a equivalência dos diferentes GCs se baseia na sua acção anti-inflamatória e não em diferentes aspectos do metabolismo humano. Uma vez que o cortisol afeta quase 20% do genoma humano, são esperadas respostas diversas de diferentes GCs em vários tecidos. Corroborar esta percepção é o facto de a administração de dexametasona a doentes com CAH ter demonstrado deterioração dos índices de resistência à insulina em comparação com outros GCs (44). Adicionalmente, a administração de 2, 5–7, 5 mg de prednisolona, uma dose considerada normal, exerce um impacto negativo de longa data no metabolismo ósseo (45). Assim, a hidrocortisona deve ser considerada a melhor forma de tratamento em casos de terapêutica de suplementação com GC. O advento da fórmula de hidrocortisona recentemente sintetizada com uma pílula por dia e seus resultados iniciais positivos em pacientes com CAH mostra muita promessa para o futuro (46).os doentes diagnosticados durante a infância com sinais de PP podem ser tratados com hidrocortisona com o objectivo de suprimir as hormonas supra-renais e impedir o rápido avanço da idade óssea que possa afectar a altura final. Nos doentes em que o tratamento com hidrocortisona foi iniciado 1 ano antes do início da puberdade e com idade óssea inferior a 9 anos, a altura final manteve-se dentro do potencial genético (47). Os estudos disponíveis indicam que a altura adulta se aproximou da altura-alvo esperada em doentes que foram cuidadosamente monitorizados e que cumpriram rigorosamente os planos de medicação (48-50). Um pequeno estudo recente de cinco casos (doentes com 6, 1–9, 2 anos de idade) demonstrou que uma dose ultraliberal de dexametasona é uma opção promissora para reduzir o stress endógeno e os seus efeitos. Se tal constitui uma abordagem realista e em que medida a dose administrada deve ser ainda elucidada (51). A study by Nebesio et al. comparou os efeitos hormonais e a farmacocinética da hidrocortisona, prednisolona e dexametasona em 9 doentes pré-pubertais com CCAH. Eles mostraram que a dexametasona era mais potente que as outras formas em alcançar níveis significativamente mais baixos de hormônio adrenal, sugerindo assim que a dexametasona é mais eficaz para a supressão da produção de androgênio adrenal (46). É claro que são necessários mais estudos para verificar ou rejeitar esta conclusão. Além disso, em alguns casos, concentrações elevadas de androgênio podem levar à estimulação secundária do eixo GnRH, levando à puberdade prematura. Nesse caso, pode ser prescrito um curso paralelo com um análogo GnRH se a idade óssea for significativamente superior à idade cronológica e/ou se a altura final projectada for desproporcionada em relação à altura-alvo.outra consideração importante diz respeito aos casos de deficiência concomitante de GH no caso de o tratamento com análogos GnRH e a prescrição de GH resultarem na consecução da altura-alvo (52). De acordo com as normas orientadoras actuais, a terapêutica acima descrita é cuidadosamente recomendada apenas nos casos em que a altura previsível DP é -2,25< a altura-alvo ou ainda mais baixa (53). Uma indicação alternativa para o início do tratamento com hidrocortisona é uma resposta cortisol inadequada após estimulação da ACTH (36).
o tratamento preferencial com GC em crianças é normalmente de 10-15 mg / m2 de hidrocortisona, dividida em três doses. Frequentemente, doses mais baixas também se revelaram eficazes, a partir de 6 mg/m2 / dia (34). A sobredosagem deve ser evitada, considerando-se que pode resultar em fraco crescimento, bem como Cushingoid características. Padrão de crescimento Regular, uma idade óssea compatível com a idade cronológica, e ausência de obesidade central também pode servir como índices clínicos para o manejo adequado. No que diz respeito ao perfil bioquímico/ hormonal, os níveis de androstenediona e testosterona nos intervalos médio-superior para a idade óssea são considerados melhores marcadores da terapêutica adequada de substituição de GC em crianças com NCAH. No entanto, os níveis de testosterona suprimidos foram encontrados em 10% dos doentes com NCCAH, enquanto outros 28% dos doentes apresentaram concentrações aumentadas de testosterona, sendo este fenómeno possivelmente atribuível à variabilidade da hidrocortisona (54, 55). Os valores de DHEAS são reduzidos com doses muito pequenas, enquanto que a supressão de 17 OHP e níveis de progesterona requer doses muito elevadas de GC. Note–se que 17 valores de OHP são cruciais para o diagnóstico, mas não são úteis durante o acompanhamento. Note-se que a colheita de amostras de sangue para avaliação hormonal deve ser realizada sem interrupção da terapêutica. No entanto, devido à perplexidade da doença e à sua natureza multifacetada, não existem orientações específicas para o momento das alterações do regime ou para a cessação da terapêutica com glucocorticóides em crianças.tratamento durante a adolescência doentes tratados desde a infância até ao estabelecimento do padrão menstrual normal nas raparigas da NCCAH, recomenda-se muito a continuação do SCG que começou durante a infância (56). Contudo, os doentes adolescentes não demonstram frequentemente o cumprimento suficiente da administração crónica de medicamentos e omitem frequentemente doses. Adicionalmente, durante a puberdade, a semi-vida da hidrocortisona diminui 50% como resultado do aumento dos níveis de IGF-1, o que diminui a actividade do 11ßOHSD, bem como devido ao aumento da depuração do cortisol resultante da amplificação da taxa de filtração glomerular (57). Na jovem fêmea, 2 anos após a menarca e se os ciclos ovulatórios normais foram registrados, uma abordagem centrada no paciente para os sintomas hiperandrogênicos que podem aparecer é altamente recomendada.se existirem resultados hiperandrogénicos graves, tais como hirsutismo, acne e/ou oligomenorreia, será considerada a continuação do tratamento. É importante lembrar as frágeis sensibilidades de um jovem adolescente, que serão seriamente danificadas pelos sinais “Repelentes” do hiperandrogenismo. A combinação de sinais hiperandrogênicos, distúrbios menstruais e má qualidade de vida está bem documentada e é particularmente alta em mulheres mais jovens. Outro ponto a ser considerado é a resposta da reserva supra-renal após a estimulação da ACTH. Dado que o cortisol máximo da puberdade e das mulheres adultas após estimulação é inferior a 496 nmol/L, em casos de stress, deve ser administrado tratamento com esteróides (58). Por outro lado, se nenhum dos sintomas acima mencionados for encontrado no doente jovem, o tratamento com GC pode ser interrompido, após o que é aconselhado um acompanhamento regular.Mulheres Com primeira manifestação durante a adolescência ou sintomas Hiperandrogénicos após interrupção do tratamento, na presença de sintomas hiperandrogénicos, recomenda-se uma abordagem orientada para o doente, focando-se nas principais queixas do doente. Isto é muito melhor do que um modus operandi universal geral, uma vez que, com a última abordagem, o jovem paciente ficará desapontado, o que leva à interrupção do tratamento e, como resultado, a resultados adversos para a saúde na idade adulta. Em casos com hirsutismo grave, um curso de 6 a 12 meses com pílulas contraceptivas orais (OCPs) constitui um procedimento razoável dado os efeitos benéficos dos OCPs na produção hepática de SHBG e a diminuição da libertação androgênica do ovário. Espera-se uma melhoria clínica após, pelo menos, 2-3 meses de início da OCPs, sendo altamente recomendada a utilização concomitante de um progestagénio com propriedades androgénicas mínimas. Se os DPOC falharem com a aproximação de primeira linha, podem ser adicionados ao tratamento antiandrogénios (espironolactona, flutamida, bicalutamida, acetato de ciproterona e finasterida). Na nossa experiência, a administração de bicalutamida atingiu uma melhoria significativa em casos de acne grave, mas não foram obtidos resultados semelhantes em casos com hirsutismo grave. Abordagens cosméticas, como aplicação laser e depilatórios também podem ser sugeridas para mulheres queixando-se de crescimento excessivo ou indesejado de cabelo ou perda de cabelo do couro cabeludo (alopecia androgênica) (32, 34, 57).
em doentes com secreção inadequada de cortisol após estimulação, ou se os AOC e os antiandrogénios não puderem ser tolerados, recomenda-se altamente um esquema de tratamento com Glcs (57). Dados de New et al. indicar que o fluxo menstrual irregular e a acne são revertidos no prazo de 3 meses após o início da terapêutica com glucocorticóides, enquanto o hirsutismo requer quase 30 meses (59). Em contraste com a infância, na adolescência, os esteróides de acção mais prolongada são frequentemente utilizados e são recomendados regimes de 5 mg de prednisolona ou 0, 25 mg de dexametasona (53). No entanto, no mundo real, os dados clínicos mostraram uma variedade de regimes diferentes aplicados na gestão da NCCAH (41).o objectivo principal do tratamento deve ser o alívio do doente dos sintomas hiperandrogénicos. Assim, entre os indivíduos com NCCAH diagnosticados com base unicamente nos resultados laboratoriais, as características clínicas devem orientar as recomendações de gestão, uma vez que os resultados hormonais e moleculares não prevêem necessariamente a gravidade clínica. De acordo com as normas orientadoras da Sociedade Endócrina, aos doentes com NCCAH deve ser dada a opção de descontinuar a terapêutica com GC quando os sintomas desaparecem (60). É claro que estes doentes não devem ser perdidos no seguimento, enquanto o tratamento deve ser reiniciado no caso de recorrência dos sintomas. Além disso, em caso de interrupção, os doentes devem ser informados sobre a possibilidade de infertilidade e devem ser encorajados a procurar aconselhamento médico se desejarem engravidar (19). De notar, a transferência apropriada do paciente da pediatria para o endocrinologista adulto deve ser realizada, de forma otimizada após 1 ano de monitorização sincronizada (53, 61).
NCCAH e Reprodução
Infertilidade
Infertilidade é comumente relatados em formas clássicas de CAH, que é principalmente devido a distúrbios menstruais, anovulação crônica e deformidades anatômicas. A taxa de natalidade foi estimada em 17% em comparação com 65% da população controladora (62). Entretanto, os dados relativos à fertilidade em mulheres com NCCAH foram recentemente avaliados em detalhe e a incidência estimada de infertilidade é de 11% entre as mulheres com NCCAH, ou seja, relativamente mais suave do que na CAH, e em muitos casos é fácil de resolver. Bidet et al. avaliou a fertilidade em 190 mulheres que sofriam de NCCAH, 95 das quais queriam engravidar. Nesta população, foram notificadas 187 gravidezes em 85 mulheres, o que resultou em 141 nascimentos em 82 indivíduos. Deve ser destacado que 99 da gravidez (52.9%) ocorreu antes do diagnóstico de NCCAH, três delas com a aplicação de protocolos de indução da ovulação, sendo o restante espontânea, enquanto 88 gravidez ocorreu post NCCAH diagnóstico. Na grande maioria delas, a gravidez desenvolveu-se após a instituição da terapêutica com hidrocortisona, enquanto em 11 mulheres ocorreu espontaneamente (63, 64). Num outro relatório de 22 mulheres observadas com NCAH desejosas de engravidar, 12 gravidezes se seguiram com prednisona (65).aborto espontâneo na coorte de Bidet e outros. a taxa de abortos foi de 6, 5 e 26, 3% em doentes tratados com GCs e doentes não tratados, respectivamente (64). O resultado da gravidez pode até ser bem sucedido sem qualquer tratamento glucocorticóide nos casos em que a NCCAH ainda não foi diagnosticada, como também relatado em um caso relatado por Cuhaci et al. No entanto, o mesmo casal teve dois abortos espontâneos e relatou subfertilidade após esta primeira gravidez (66). No que diz respeito à possibilidade de gravidezes a termo, tal como avaliado por Bidet et al., não difere significativamente do resto da população (15).planeamento da fertilidade para mulheres com hiperandrogenismo sintomático ou com infertilidade notificada mas que desejem engravidar, recomenda-se vivamente a terapêutica com CG. Durante este período, os glucocorticóides utilizados são a prednisona ou a hidrocortisona. A ação mais importante, porque irá orientar os passos subsequentes, é o teste genético dos futuros pais. As fêmeas com NCCAH que desejam engravidar devem estar cientes do risco de dar à luz uma criança com a forma clássica da doença. O resultado da gravidez entre mulheres com NCCAH, e mais especificamente a incidência de crianças nascidas com CCAH, é estimado em estudos recentes entre 1,5 e 2,5%, com NCCAH em cerca de 15% (36, 64). É claro que esta frequência depende da população de referência, uma incidência mais elevada ocorrendo em populações com altas taxas de casamentos inter-conjugais (36). As chances previstas de os pais darem à luz uma criança com CAH é de 1 em 120 para o genótipo paterno desconhecido, zero se o pai não tem mutação relevante, e 1 em 4 se ele tem uma mutação heterozigótica (19). Como resultado, o teste genético dos parceiros destas mulheres é essencial para avaliar o risco de dar à luz uma criança com a forma clássica de CAH (64, 67).
no caso de a futura mãe ter duas mutações NC( Tabela 1), não há necessidade absoluta de testes genéticos no futuro pai, uma vez que, se ele for heterozigótico para as mutações CYP12A2, a descendência corre o risco de desenvolver a forma não-clássica da doença no futuro. No entanto, deve-se notar que o estatuto de transportador é de 2-15% em populações diferentes e metade destes indivíduos possuem uma mutação grave. Além disso, as recomendações definitivas relativas a situações em que os ensaios genéticos não são necessários são difíceis, dada a correlação imperfeita genótipo-fenótipo, particularmente para mutações mais brandas . Por exemplo, a mutação P30L, mais frequentemente associada à NCCAH, também ocorre em doentes com CAH clássica . Um grande estudo europeu multicêntrico mostrou recentemente que este foi o caso em 78% dos doentes . Neste estudo, a mutação ligeira V218L foi associada a mutação clássica em vez de NCCAH em 30% dos casos. Assim, todos os doentes com NCCAH devem receber aconselhamento genético e avaliação molecular de parceiros reprodutivos.
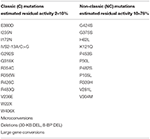
Tabela 1. Mutações do CYP21A2 com actividade enzimática residual mínima (C) ou moderada (NC) (14, 68, 69).
por contraste, no caso de a mãe potencial ser um heterozigoto composto com uma mutação grave (C) e NC, então o teste genético do futuro pai é obrigatório. Devemos ter em mente que os dados provenientes de estudos que utilizam imunoensaios ou os procedimentos LC-MS/MS mais precisos, destacam consistentemente a incapacidade de excluir de forma confiável heterozigosidade usando valores basais ou ACTH estimulados 17 OHP, devido à sobreposição significativa com o intervalo normal. Pode-se sugerir o uso de um teste Sinacteno, e se não for compatível com heterozigosidade (soma de valores basais e de pico estimulado 17 valores OHP < 1,5 nmol/L), então o teste de DNA poderia ser evitado. No entanto, devemos ser muito cautelosos na interpretação deste teste, uma vez que não foi validado noutros estudos. Portanto, o uso de ACTH estimulado 21-deoxycortisol individualmente ou como parte de um esteróide relação ou esteróides perfil, pode facilitar a identificação bioquímica de heterozigotos no futuro, particularmente como LC-MS/MS torna-se mais amplamente disponível . Se o futuro pai carrega uma mutação NC, então nada mais é necessário. Inversamente, se ele é um portador de uma mutação clássica, a questão relativa ao tratamento pré-natal do feto vai surgir. Todas as combinações possíveis de genótipos e os procedimentos sugeridos são apresentados na Tabela 2.

Tabela 2. Planeamento dos procedimentos sugeridos durante a gravidez com base no genótipo potencial dos pais.
tratamento pré-natal de CAH
o objetivo do tratamento pré-natal de CAH é a prevenção da virilização genital do feto, mas também aliviar o estresse sentido pelos pais que são susceptíveis de ter um filho com genitália ambígua (70). A dexametasona é utilizada devido à sua capacidade de atravessar a placenta e porque não é desactivada a partir da placenta 11β Oh esteróide desidrogenase e liga-se apenas minimamente à globulina ligante de cortisol no sangue da mãe (CBG). A dexametasona, ao suprimir a secreção de ACTH fetal, diminui a produção de androgénios fetais elevada. O tratamento deve ser interrompido caso o cariótipo ou a análise de ADN revele, respectivamente, um macho ou uma fêmea não afectada (71). No entanto, embora a virilização genital fetal já tenha começado em 6-7 semanas após a concepção, biópsias coriônicas villous para o diagnóstico genético não pode ser obtida antes da 10ª−12ª semana. Este intervalo de tempo sugere que todas as gravidezes em risco de virilização de CAH devem ser tratadas, apesar de apenas 1 em cada 8 fetos ser afetado e feminino (19, 34, 70).se esta exposição do feto à dexametasona para medidas preventivas é clinicamente e eticamente aceitável continua a ser controversa (70). Estudos em animais demonstraram que a dexametasona do primeiro trimestre diminui o peso à nascença, afecta a função das células beta pancreáticas renais e o desenvolvimento cerebral, e predispõe para ansiedade, hipertensão e hiperglicemia na idade adulta (70). Além disso, 10-20% das mulheres grávidas que utilizam o tratamento com dexametasona durante a gravidez queixam-se de aumento de peso, aumento do apetite, alterações do humor, insónia, edemas, hipertensão, hiperglicemia e estrias. No entanto, New et al. detectou uma pontuação Prader inferior à média em fetos tratados com dexametasona pré-natal, mas sem diferença nos resultados a longo prazo (72). Além disso, em uma revisão de Merce Fernandez-Balsells et al. a dexametasona demonstrou estar associada à redução da virilização do feto sem efeitos adversos maternos ou fetais significativos (63). No entanto, os autores da referida revisão, bem como as Diretrizes consecutivas da Sociedade Endócrina, apontam que, devido ao pequeno tamanho das amostras em todo o corpo da literatura, o assunto permanece incerto e é claramente necessária uma investigação mais aprofundada.
a decisão de iniciar o tratamento deve ser tomada apenas em grandes centros com uma equipe experiente e protocolos aprovados por conselhos de revisão institucionais e com base nos valores e preferências da família e com seu consentimento informado por escrito como pré-requisito (19, 63). Vários especialistas no campo indicam que um valor mais elevado deve ser colocado na prevenção da exposição pré-natal desnecessária de mãe e feto à dexametasona, em vez de impor o pedágio emocional de genitália ambígua aos pais e pacientes. Mais importante ainda, deve ser esclarecido aos pais que a administração de dexametasona não modifica o estado do paciente, mas é direcionada para reduzir a necessidade de cirurgia ao invés de preservar a vida ou a capacidade intelectual. Eles também devem saber que a probabilidade de que seu filho vai sofrer da forma clássica da doença é alta, apesar do tratamento. Mais crucialmente, entretanto, a exposição de um organismo jovem a um GC muito potente durante um período particularmente sensível de programação e crescimento fetal, que pode muito bem se revelar inútil no caso de um feto com o cariótipo XY, não é atualmente apoiada por dados robustos e inquestionáveis.idealmente, deve haver um diagnóstico precoce, realizado através de uma técnica não invasiva e antes do início da virilização genital do feto. Trabalhando nesta direção, novos estudos apontam para o uso de DNA fetal livre de células obtido a partir de plasma materno como um método promissor que permitirá a determinação do sexo fetal e o diagnóstico de CAH em uma idade gestacional precoce (<9 semanas). Se este procedimento for amplamente implementado na prática clínica, o tratamento pré-natal desnecessário de dexametasona será evitado (73).tratamento durante a gravidez
durante a gravidez, as mulheres com NCCAH devem ser preferencialmente tratadas com hidrocortisona, não só porque a gravidez é tipicamente acompanhada por níveis elevados de stress, mas também como uma medida preventiva contra a elevada incidência acima referida de abortos espontâneos. Esta forma de glucocorticóide é favorecida devido à sua metabolização pela enzima placentária 11β OH esteróide desidrogenase II, que impede o glucocorticóide de ter qualquer efeito sobre o feto (71). Na altura da concepção, a dose de hidrocortisona deve ser aumentada para 20-25 mg/dia e as modificações da dose são efectuadas a cada 6-8 semanas com o objectivo de manter os níveis de testosterona nos níveis normais superiores do trimestre da gravidez (74). Como exemplo, os valores de testosterona e D4 de um paciente com NCCAH desde a concepção até 3 anos após o parto de dois gêmeos saudáveis são mostrados (Figura 1).
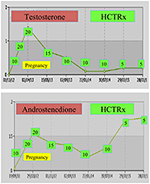
Figura 1. Níveis de testosterona e andostenediona e subsequente dose de hidrocortisona (HCTRx), desde a concepção até 3 anos após o parto, administrada a um doente com NCCAH.
outras questões especiais
controlo do Stress na NCCAH
durante o stress grave com risco de vida, cirurgia ou doença grave, os doentes com NCCA que são tratados com glucocorticóides podem necessitar de doses maiores ou mais frequentes de glucocorticóides, dado que a sua função adrenal é iatrogenicamente suprimida. Por conseguinte, é fundamental educar os pais de crianças pequenas, bem como reeducar os doentes sobre a sua transição para os cuidados de adultos, sobre a dosagem do stress. Para os doentes com NCCAH que não estejam a ser tratados com Glasgow ou em caso de descontinuação, deve avaliar-se a resposta do cortisol ao ACTH. Quase um terço dos doentes com NCCAH responde inadequadamente à estimulação (15, 75). Para os que respondem normalmente à estimulação da ACTH, não é recomendado tratamento com doses de stress (19). A terapêutica mineralocorticóide não é necessária em nenhum dos casos, uma vez que estes doentes têm uma secreção normal de aldosterona e não desenvolvem desperdício de sal (58). De notar que, em caso de hipertiroidismo ou de aumento do tratamento com levotiroxina, a depuração do cortisol está aumentada e pode ocorrer uma crise adrenal (58, 76).
aspectos Psicobiológicos na NCCAH
Meyer-Bahlburg et al. em vários estudos relataram diminuição do perfil psicológico entre os doentes com NCCAH devido a deficiência de 21OH. Mais especificamente, as mulheres afetadas tiveram uma maior pontuação de masculinização/desfeminização em várias medidas de comportamento relacionado com o gênero quando comparado com as mulheres de controle normal, embora marcadamente menos do que em mulheres com CAH clássica. Eles também tinham aumentado significativamente a orientação bissexual ou homossexual. Entrevistas clínicas-qualitativas retrospectivas com estas mulheres revelaram uma história de desconforto e estresse social relacionados com suas experiências pré-tratamento com sinais androgênicos dependentes, tais como acne, hirsutismo e dificuldades de concepção (59, 77, 78). Similarmente, Arlt et al. mostrou que o estado de saúde subjetivo foi significativamente prejudicado em todos os oito domínios de um levantamento de saúde de curto prazo, com as diferenças mais proeminentes, em comparação com a idade e os controles combinados com o Sexo, relacionados com os domínios saúde geral, vitalidade e limitações de papel devido a problemas emocionais. O questionário da escala de ansiedade e depressão Hospitalar (HADS) também revelou um aumento na pontuação de ansiedade (41). Tendo em conta estes achados, os parâmetros psicológicos para orientar a terapia devem ser considerados em mulheres com NCCAH e, no contexto da abordagem orientada para o paciente, um diagnóstico psicológico e apoio deve ser oferecido.
conclusões
NCCAH é considerada como a forma mais frequente e mais suave de CAH devido à retenção de 20-50% de actividade enzimática. A maioria dos doentes pode procurar aconselhamento médico em qualquer fase da sua vida devido a sintomas relacionados com o excesso de androgénios. Estes incluem pubarche prematuro, crescimento rápido, hirsutismo, acne, irregularidades menstruais, ou infertilidade, bem como muitas outras manifestações menos proeminentes. São recomendados valores basais iniciais de 17 OHP no início da manhã, como um bom teste de rastreio inicial e uma avaliação adicional com estimulação da ACTH e, no caso de resultados limítrofes, testes genéticos. Como regra geral, os níveis de androstenediona e não 17 OHP devem ser usados durante o acompanhamento.a decisão de tratamento deve basear-se na avaliação dos factos e seguir uma abordagem individualizada. O tratamento nem sempre é indicado a menos que o doente seja sintomático, por exemplo, crianças com início precoce e progressão rápida dos pêlos púbicos e corporais, crescimento rápido e/ou avanço esquelético, ou mulheres com oligomenorreia, acne, hirsutismo, infertilidade, ou uma combinação destes e de outros dos sintomas acima referidos. O aconselhamento genético é fortemente aconselhado em mulheres NCCAH que desejam engravidar, bem como genotipagem do Pai. O tratamento global do doente inclui adicionalmente o tratamento das prováveis complicações da terapêutica com glucocorticóides ou manifestações relacionadas com o metabolismo da doença. Por último, dado que muitas questões terapêuticas relacionadas com a gestão adequada destes doentes ainda não foram esclarecidas, é muito importante que o médico assistente mantenha-se actualizado sobre todos os desenvolvimentos neste domínio e integre os novos dados na sua prática clínica. É certo que são essenciais mais estudos clínicos nesta área.para resumir, para a mulher NCC, a abordagem ideal é feita sob medida, incorporando uma transição suave de sua gestão, uma vez que ela é encaminhada da pediatria para o endocrinologista adulto, juntamente com tratamento orientado aos sintomas que irá acompanhá-la ao longo de sua vida. Tendo em conta os múltiplos factores que afectam o sistema hiperandrogénico, é aconselhável incentivar os doentes a adoptarem um estilo de vida mais saudável, melhorando os seus hábitos alimentares, aumentando o exercício e visando a redução de peso. Além disso, em certos casos, o apoio psicológico é frequentemente benéfico. Além disso, especialistas em áreas envolvidas no tratamento desta doença, como dermatologistas, ginecologistas e psicólogos, precisam de uma compreensão profunda sobre a gestão de casos suspeitos ou já diagnosticados de NCCAH. Para concluir, vamos ter em mente a citação perspicaz da astrónoma Americana Vera Cooper Rubin: “a Ciência progride melhor quando as observações nos obrigam a alterar os nossos preconceitos.”
contribuições do autor
todos os autores listados fizeram uma contribuição substancial, direta e intelectual para a obra, e aprovou-a para publicação.
Declaração de conflito de interesses
os autores declaram que a investigação foi realizada na ausência de quaisquer relações comerciais ou financeiras que possam ser interpretadas como um potencial conflito de interesses.
1. Speiser PW, White PC. Hiperplasia adrenal congénita. N Engl J Med. (2003) 349:776–88. doi: 10.1056/NEJMra021561
PubMed Resumo | CrossRef Texto Completo | Google Scholar
2. Turcu AF, Auchus RJ. Adrenal steroidogenesis and congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. (2015) 44:275–96. doi: 10.1016/j.ecl.2015.02.002
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Speiser PW. Nonclassic adrenal hyperplasia. Rev Endocr Metab Disord. (2009) 10:77–82. doi: 10.1007/s11154-008-9097-x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Gidlöf S, Falhammar H, Thilén a, von Döbeln U, Ritzén M, Wedell A, et al. Cem anos de hiperplasia adrenal congênita na Suécia: um estudo retrospectivo baseado em coorte populacional. Lancet Diabetes Endocrinol. (2013) 1:35–42. doi: 10.1016/S2213-8587(13)70007-X
PubMed Resumo | CrossRef Texto Completo | Google Scholar
5. Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, New MI. Alta frequência de deficiência em esteroides 21-hidroxilase não Classe. Am J Hum Genet. (1985) 37:650–67.
PubMed Abstract | Google Scholar
6. Wilson RC, Nimkarn S, Dumic M, Obeid J, Azar MR, Azar M, et al. Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Mol Genet Metab. (2007) 90:414–21. doi: 10.1016/j.ymgme.2006.12.005
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Miller WL. Clinical review 54: genetics, diagnosis, and management of 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1994) 78:241–6. doi: 10.1210/jcem.78.2.8106606
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Witchel SF, Azziz R. Nonclassic congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2010) 2010:625105. doi: 10.1186/1687-9856-2010-625105
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Wilson RC, Mercado AB, Cheng KC, New MI. Deficiência em esteroides 21-hidroxilase: o genótipo pode não prever fenótipo. J Clin Endocrinol Metab. (1995) 80:2322–9. doi: 10.1210 / jc.80.8.2322
CrossRef Texto Completo | Google Scholar
10. PC branco. Rastreio Neonatal para hiperplasia adrenal congénita. Nat Rev Endocrinol. (2009) 5:490–8. doi: 10.1038 / nrendo.2009.148
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Kohn B, Levine LS, Pollack MS, Pang S, Lorenzen F, Levy D, et al. Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. (1982) 55:817–27. doi: 10.1210/jcem-55-5-817
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Moran C, Azziz R, Carmina e, Dewailly D, Fruzzetti F, Ibañez L, et al. 21-Hydroxylase-deficient nonclassic adrenal hyperplasia is a progressive disorder: a multicenter study. Am J Obstet Gynecol. (2000) 183:1468–74. doi: 10.1067 / mob.2000.108020
PubMed Abstract / CrossRef Full Text/Google Scholar
13. Azziz R, Hincapie LA, Knochenhauer ES, Dewailly D, Fox l, Boots LR. Screening for 21-hydroxylase-deficient nonclassic adrenal hyperplasia among hyperandrogenic women: a prospective study. Fertilizante. (1999) 72:915–25. doi: 10.1016/S0015-0282(99)00383-0
PubMed Resumo | CrossRef Texto Completo | Google Scholar
14. Livadas S, Dracopoulou M, Dastamani a, Sertedaki a, Maniati-Christidi M, Magiakou A-M, et al. O espectro de resultados clínicos, hormonais e moleculares em 280 indivíduos com hiperplasia adrenal congénita nãoclássica causada por mutações do gene CYP21A2. Clin Endocrinol (Oxf). (2015) 82:543–9. doi: 10.1111 / cen.12543
PubMed Resumo | CrossRef Texto Completo | Google Scholar
15. Bidet M, Bellanné-Chantelot C, Galand-Portier m-B, Tardy V, Billaud L, Laborde K, et al. Caracterização clínica e molecular de uma coorte de 161 mulheres não relacionadas com hiperplasia adrenal congénita nãoclássica devido a deficiência de 21-hidroxilase e 330 membros da família. J Clin Endocrinol Metab. (2009) 94:1570–8. doi: 10.1210 / jc.2008-1582
PubMed Resumo | CrossRef Texto Completo | Google Scholar
16. Escobar-Morreale HF, Sanchón R, San Millán JL. Um estudo prospectivo da prevalência de hiperplasia adrenal congénita não-clássica entre as mulheres que apresentam sintomas e sinais hiperandrogénicos. J Clin Endocrinol Metab. (2008) 93:527–33. doi: 10.1210 / jc.2007-2053
PubMed Resumo | CrossRef Texto Completo | Google Scholar
17. Dewailly D, Vantyghem-Haudiquet MC, Sainsard C, Buvat J, Cappoen JP, Ardaens K, et al. Fenótipos clínicos e biológicos em deficiência de 21-hidroxilase de início tardio. J Clin Endocrinol Metab. (1986) 63:418–23. doi: 10.1210/jcem-63-2-418
PubMed Resumo | CrossRef Texto Completo | Google Scholar
18. Gönç EN, Ozön ZA, Alikaşifoglu a, Engiz o, Bulum B, Kandemir N. (2011). Is basal serum 17-OH progesterone a reliable parameter to predict nonclassical congenital adrenal hyperplasia in premature adrenarche? Turk J Pediatr. 53:274–80.
PubMed Abstract/Google Scholar
19. Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endócrino society clinical practice guideline. J Clin Endocrinol Metab. (2010) 95:4133–60. doi: 10.1210 / jc.2009-2631
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Dacou-Voutetakis C, Dracopoulou M. High incidence of molecular defects of the CYP21 gene in patients with premature adrenarche. J Clin Endocrinol Metab. (1999) 84:1570–4. doi: 10.1210/jc.84.5.1570
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Ambroziak y, Kepczynska-eventos I, Kurylović, Małunowicz EM, Wójcicka, Miskiewicz P, et al. O diagnóstico de hiperplasia congênita nonclassic do córtex adrenal deve-se à deficiência de 21-hidroxilase, com base no soro basal ou na estimulação pós-activa de 17-hidroxiprogesterona, pode levar ao diagnóstico falso-positivo. Clin Endocrinol (Oxf). (2016) 84:23–9. doi: 10.1111 / preços.12935
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Ambroziak y, Kepczynska-eventos I, Kurylovich, Wysłouch-Cieszyńska a, Małunowicz EM, Bartoszewicz, et al. LC-MS/MS improves screening towards 21-hydroxylase deficiency. Gynecol É Um Endocrinol. (2015) 31:296–300. doi: 10.3109/09513590.2014.994599
PubMed Abstract | CrossRef Full Text | Google Scholar
23. BBC, CC, Goldman mm, Zhang K, Clark-N, Wrightz RE Welt CK. Medição simultânea de treze hormônios esteróides em mulheres com síndrome de ovário policístico e mulheres de controle usando cromatografia líquida-espectrometria de massa tandem. PLoS Um. (2014) 9:e93805. doi: 10.1371 / journal.pone.0093805
PubMed Resumo | CrossRef Texto Completo | Google Scholar
24. Ray JA, Kushnir MM, Yost RA, Rockwood AL, Wayne Meikle A. Performance enhancement in the measurement of 5 endogenous steroids by LC-MS/MS combined with differential ion mobility spectrometry. Clin Chim Acta. (2015) 438:330–6. doi: 10.1016 / j. cca.2014.07.036
PubMed Abstract / CrossRef Full Text/Google Scholar
25. Stoupa a, González-Briceño l, Pinto G, Samara-Boustani D, Thalassinos C, Flechtner I, et al. Inadequate cortisol response to the tetracosactide (Synacthen®) test in non-classic congenital adrenal hyperplasia: an exception to the rule? Ant Res Paediatr. (2015) 83:262–7. doi: 10.1159/000369901
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. (1992) 90:584–95. doi: 10.1172/JCI115897
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Temeck JW, Pang SY, Nelson C, New MI. Defeitos genéticos da esteroidogénese em pubarche prematuro. J Clin Endocrinol Metab. (1987) 64:609–17. doi: 10.1210/jcem-64-3-609
PubMed Resumo | CrossRef Texto Completo | Google Scholar
28. Voutilainen R, Jääskeläinen J. Premature adrenarche: etiology, clinical findings, and consequences. J Bioquímica De Esteróides Mol Biol. (2015) 145:226–36. doi: 10.1016 / j. jsbmb.2014.06.004
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Oberfield SE, Sopher AB, Gerken AT. Approach to the girl with early onset of pubic hair. J Clin Endocrinol Metab. (2011) 96:1610–22. doi: 10.1210/jc.2011-0225
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Janus D, Wojcik M, Malunowicz E, Starzyk JB. Um caso de adesão labial recorrente numa criança de 15 meses com hiperplasia adrenal congénita assintomática não clássica devido a deficiência de 21-hidroxilase. Pediatra Endocrinol Metab. (2012) 25:1017–21. doi: 10.1515/jpem-2012-0190
PubMed Resumo | CrossRef Texto Completo | Google Scholar
31. Gopal-Kothandapani JS, Petkar a, O’Shea e, Banerjee I. Perianal hair as an unusual presentation of non-classical congénite adrenal hyperplasia. BMJ processo Rep. (2013) 2013:bcr20130123. doi: 10.1136/bcr-2013-010123
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Falhammar H, Nordenström A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine. (2015) 50:32–50. doi: 10.1007/s12020-015-0656-0
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Chrousos GP, Loriaux DL, Mann DL, Cutler GB. Late-onset 21-hydroxylase deficiency mimicking idiopathic hirsutism or polycystic ovarian disease. Ann Intern Med. (1982) 96:143–8. doi: 10.7326/0003-4819-96-2-143
CrossRef Full Text | Google Scholar
34. Witchel SF. Non-classic congenital adrenal hyperplasia. Steroids. (2013) 78:747–50. doi: 10.1016/j.steroids.2013.04.010
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Carmina E, Rosato F, Jannì A, Rizzo M, Longo RA. Extensive clinical experience: relative prevalence of different androgen excess disorders in 950 women referred because of clinical hyperandrogenism. J Clin Endocrinol Metab. (2006) 91:2–6. doi: 10.1210/jc.2005-1457
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Moran C, Azziz R, Weintrob N, Witchel SF, Rohmer V, Dewailly D, et al. Resultados reprodutivos de mulheres com hiperplasia adrenal não clássica com deficiência em 21-hidroxilase. J Clin Endocrinol Metab. (2006) 91:3451–6. doi: 10.1210 / jc.2006-0062
PubMed Resumo | CrossRef Texto Completo | Google Scholar
37. Levin JH, Carmina e, Lobo RA. A secreção inapropriada de gonadotropina em doentes com síndrome do ovário policístico é semelhante à de doentes com hiperplasia adrenal congénita de início adulto? Fertilizante. (1991) 56:635–40. doi: 10.1016/S0015-0282(16)54592-0
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Carmina E, Lobo RA. Ovarian suppression reduces clinical and endocrine expression of late-onset congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Fertil Steril. (1994) 62:738–43. doi: 10.1016/S0015-0282(16)56998-2
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Ludwig E. Classification of the types of androgenetic alopecia (common baldness) occurring in the female sex. Br J Dermatol. (1977) 97:247–54. doi: 10.1111 / J. 1365-2133. 1977.tb15179.x
PubMed Resumo | CrossRef Texto Completo | Google Scholar
40. Trakakis E, Papadavid E, Dalamaga M, Koumaki D, Stavrianeas N, Rigopoulos D, et al. Prevalência de hiperplasia adrenal congénita não-clássica devido a deficiência de 21-hidroxilase em mulheres gregas com acne: um estudo transversal baseado em hospitais. J Eur Acad Dermatol Venereol. (2013) 27:1448–51. doi: 10.1111 / J. 1468-3083. 2012. 04613.x
PubMed Resumo | CrossRef Texto Completo | Google Scholar
41. Arlt W, Willis DS, Wild SH, Krone N, Doherty EJ, Hahner S, et al. Estado de saúde dos adultos com hiperplasia adrenal congénita: um estudo de coorte de 203 doentes. J Clin Endocrinol Metab. (2010) 95:5110–21. doi: 10.1210 / jc.2010-0917
PubMed Resumo | CrossRef Texto Completo | Google Scholar
42. Saygili F, Oge a, Yilmaz C. hiperinsulinemia e insensibilidade à insulina em mulheres com hiperplasia adrenal congénita não clássica devido a deficiência de 21-hidroxilase: a relação entre os níveis séricos de leptina e a hiperinsulinemia crónica. Horm Res. (2005) 63:270-4. doi: 10.1159/000086363
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Speiser PW, Serrat J, New MI, Gertner JM. Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1992) 75:1421–4. doi: 10.1210/jc.75.6.1421
PubMed Abstract | CrossRef Full Text | Google Scholar
44. Han TS, Stimson RH, Rees DA, Krone N, Willis DS, Conway GS, et al. Regime terapêutico com glucocorticóides e resultados para a saúde em adultos com hiperplasia adrenal congénita. Clin Endocrinol. (2013) 78:197–203. doi: 10.1111 / cen.12045
PubMed Resumo | CrossRef Texto Completo | Google Scholar
45. Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. utilização de corticosteróides orais e risco de fracturas. J Bone Miner Res. (2000) 15:993-1000. doi: 10.1359 / jbmr.2000.15.6.993
PubMed Abstract | CrossRef Full Text | Google Scholar
46. Nebesio TD, Renbarger JL, Nabhan ZM, Ross SE, Slaven JE, Li L, et al. Differential effects of hydrocortisone, prednisone, and dexamethasone on hormonal and pharmacokinetic profiles: a pilot study in children with congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2016) 2016:17. doi: 10.1186/s13633-016-0035-5
PubMed Resumo | CrossRef Texto Completo | Google Scholar
47. Weintrob n, Dickerman Z, Sprecher e, Galatzer a, Pertzelan A. Non-classical 21-hydroxylase deficiency in infancy and childhood: the effect of time of initiation of therapy on puberty and final height. Eur J Endocrinol. (1997) 136:188–95. doi: 10.1530/eje.0.1360188
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Bonfig W, Bechtold S, Schmidt H, Knorr D, Schwarz HP. Reduced final height outcome in congenital adrenal hyperplasia under prednisone treatment: deceleration of growth velocity during puberty. J Clin Endocrinol Metab. (2007) 92:1635–9. doi: 10.1210/jc.2006-2109
PubMed Abstract | CrossRef Full Text | Google Scholar
49. Dörr HG. Crescimento em doentes com hiperplasia adrenal congénita clássica devido a deficiência de 21-hidroxilase. Horm Res. (2007) 68(Suppl 5):93-9. doi: 10.1159/000110587
PubMed Resumo | CrossRef Texto Completo | Google Scholar
50. Hoepffner W, Kaufhold a, Willgerodt H, Keller E. doentes com hiperplasia adrenal congénita clássica devido à deficiência de 21-hidroxilase podem atingir a sua altura-alvo: a experiência de Leipzig. Horm Res. (2008) 70:42-50. doi: 10.1159/000129677
PubMed Abstract | CrossRef Full Text | Google Scholar
51. van der Kaay D, van den Akker E. Ultralow-dose dexamethasone to preserve endogenous cortisol stress response in nonclassical congenital adrenal hyperplasia: a new promising treatment. Int J Endocrinol Metab. (2014) 12:e14657. doi: 10.5812/ijem.14657
CrossRef Full Text | Google Scholar
52. Lin-Su K, Vogiatzi MG, Marshall I, Harbison MD, Macapagal MC, Betensky B, et al. Treatment with growth hormone and luteinizing hormone releasing hormone analog improves final adult height in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. (2005) 90:3318–25. doi: 10.1210/jc.2004-2128
PubMed Abstract | CrossRef Full Text | Google Scholar
53. McCann-Crosby B, Chen M-J, Lyons SK, Lin Y, Axelrad M, Dietrich JE, et al. Nonclassical congenital adrenal hyperplasia: objectivos de tratamento e transição. Pediatr Endocrinol Rev. (2014) 12:224-38.
PubMed Resumo | Google Scholar
54. Ogilvie CM, Crouch NS, Rumsby G, Creighton SM, Liao L-M, Conway GS. Hiperplasia adrenal congênita em adultos: uma revisão de problemas médicos, cirúrgicos e psicológicos. Clin Endocrinol. (2006) 64:2–11. doi: 10.1111 / J. 1365-2265. 2005. 02410.x
PubMed Abstract | CrossRef Full Text | Google Scholar
55. Mah PM, Jenkins RC, Rostami-Hodjegan A, Newell-Price J, Doane A, Ibbotson V, et al. Weight-related dosing, timing and monitoring hydrocortisone replacement therapy in patients with adrenal insufficiency. Clin Endocrinol. (2004) 61:367–75. doi: 10.1111/j.1365-2265.2004.02106.x
PubMed Abstract | CrossRef Full Text | Google Scholar
56. Diaz a, Laufer Sr. Breech LLAA do PC em A, Care AC of o e GC em AH. Menstruação em meninas e adolescentes: usando o ciclo menstrual como um sinal vital. Pediatria. (2006) 118:2245–50. doi: 10.1542 / Pediatria.2006-2481
CrossRef Texto Completo | Google Scholar
57. Merke DP, Poppas DP. Tratamento de adolescentes com hiperplasia adrenal congénita. lancet Diabetes Endocrinol. (2013) 1:341–52. doi: 10.1016/S2213-8587(13)70138-4
PubMed Abstract | CrossRef Full Text | Google Scholar
58. Verma S, Green-Golan L, VanRyzin C, Drinkard B, Mehta SP, Weise M, et al. Adrenomedullary function in patients with nonclassic congenital adrenal hyperplasia. Horm Metab Res. (2010) 42:607–12. doi: 10.1055/s-0030-1253385
PubMed Abstract | CrossRef Full Text | Google Scholar
59. New MI. Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2006) 91:4205–14. doi: 10.1210/jc.2006-1645
PubMed Abstract | CrossRef Full Text | Google Scholar
60. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2018) 103:4043–88. doi: 10.1210/jc.2018-01865
PubMed Abstract | CrossRef Full Text | Google Scholar
61. Kruse B, Riepe FG, Krone N, Bosinski HAG, Kloehn S, Partsch CJ, et al. Congenital adrenal hyperplasia – how to improve the transition from adolescence to adult life. Exp Clin Endocrinol Diabetes. (2004) 112:343–55. doi: 10.1055/s-2004-821013
PubMed Abstract | CrossRef Full Text | Google Scholar
62. Gastaud F, Bouvattier C, Duranteau L, Brauner R, Thibaud E, Kutten F, et al. Resultados sexuais e reprodutivos deficientes em mulheres com formas clássicas de hiperplasia adrenal congénita. J Clin Endocrinol Metab. (2007) 92:1391–6. doi: 10.1210 / jc.2006-1757
PubMed Resumo | CrossRef Texto Completo | Google Scholar
63. Mercè Fernández-Balsells M, Muthusamy K, Smushkin G, Lampropulos JF, Elamin MB, Abu Elnour no, et al. Utilização pré-natal de dexametasona na prevenção da virilização em gravidezes em risco de hiperplasia adrenal congénita clássica devido à deficiência de 21-hidroxilase (CYP21A2): uma revisão sistemática e meta-análises. Clin Endocrinol. (2010) 73:436–44. doi: 10.1111 / J. 1365-2265. 2010. 03826.x
CrossRef Texto Completo | Google Scholar
64. Bidet M, Bellanné-Chantelot C, Galand-Portier MB, Golmard JL, Tardy V, Morel Y, et al. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2010) 95:1182–90. doi: 10.1210/jc.2009-1383
PubMed Abstract | CrossRef Full Text | Google Scholar
65. Birnbaum MD, Rose LI. Late onset adrenocortical hydroxylase deficiencies associated with menstrual dysfunction. Obstet Gynecol. (1984) 63:445–51.
PubMed Abstract | Google Scholar
66. Cuhaci N, Aydin C, Yesilyurt A, Pinarli FA, Ersoy R, Cakir B. Nonclassical congenital adrenal hyperplasia and pregnancy. Case Rep Endocrinol. (2015) 2015:296924. doi: 10.1155/2015/296924
PubMed Abstract | CrossRef Full Text | Google Scholar
67. Deneux C, Tardy V, Anne D, Mornet E, Billaud L, Charron D, et al. Correlação fenótipo-genótipo em 56 mulheres com hiperplasia adrenal congénita nãoclássica devido a deficiência de 21-hidroxilase. J Clin Endocrinol Metab. (2001) 86:207–13. doi: 10.1210 / jcem.86.1.7131
PubMed Resumo | CrossRef Texto Completo | Google Scholar
68. Simonetti L, Bruque CD, Fernández CS, Benavides-Mori B, Delea M, Kolomenski JE, et al. CYP21A2 mutation update: Comprehensive analysis of databases and published genetic variants. Hum Mutat. (2018) 39:5–22. doi: 10.1002 / humu.23351
PubMed Abstract | CrossRef Full Text | Google Scholar
69. OMIM Entry – * 613815 – CYTOCHROME P450 FAMILY 21 SUBFAMILY A POLYPEPTIDE 2; CYP21A2.
Google Scholar
70. Miller WL, Witchel SF. Prenatal treatment of congenital adrenal hyperplasia: risks outweigh benefits. Am J Obstet Gynecol. (2013) 208:354–9. doi: 10.1016/j.ajog.2012.10.885
PubMed Abstract | CrossRef Full Text | Google Scholar
71. Lekarev O, New MI. Adrenal disease in pregnancy. Best Pract Res Clin Endocrinol Metab. (2011) 25:959–73. doi: 10.1016/j.beem.2011.08.004
PubMed Abstract | CrossRef Full Text | Google Scholar
72. New MI, Abraham M, Yuen T, Lekarev O. An update on prenatal diagnosis and treatment of congenital adrenal hyperplasia. Semin Reprod Med. (2012) 30:396–9. doi: 10.1055/s-0032-1324723
PubMed Abstract | CrossRef Full Text | Google Scholar
73. Kazmi D, Bailey J, Yau M, Abu-Amer W, Kumar A, Low M, et al. New developments in prenatal diagnosis of congenital adrenal hyperplasia. J Steroid Biochem Mol Biol. (2017) 165:121–3. doi: 10.1016/j.jsbmb.2016.06.016
PubMed Abstract | CrossRef Full Text | Google Scholar
74. Clayton PE, Miller WL, Oberfield SE, Ritzén EM, Sippell WG, Speiser PW, et al. Consensus statement on 21-hydroxylase deficity from the European Society for Pediátrica Endocrinology and the lawson wilkins pediatric endocrinine society. Horm Res. (2002) 58:188-95. doi: 10.1159/000065490
PubMed Resumo | CrossRef Texto Completo | Google Scholar
75. Nandagopal R, Sinaii N, Avila na, Van Ryzin C, Chen W, Finkielstain GP, et al. Análise fenotípica dos pais com hiperplasia adrenal congénita críptica não clássica: resultados em 145 famílias não relacionadas. Eur J Endocrinol. (2011) 164:977–84. doi: 10.1530/EJE-11-0019
PubMed Resumo | CrossRef Texto Completo | Google Scholar
76. Takasu N, Nakachi K, Higa H. O desenvolvimento do hipertiroidismo de Graves causou uma crise adrenal em um paciente com deficiência de 21-hidroxilase não reconhecida anteriormente. Médico Interno. (2010) 49:1395–400. doi: 10.2169 / internalmedicina.49.3573
PubMed Abstract | CrossRef Full Text | Google Scholar
77. Meyer-Bahlburg HFL, Dolezal C, Baker SW, Carlson AD, Obeid JS, New MI. Prenatal androgenization affects gender-related behavior but not gender identity in 5-12-year-old girls with congenital adrenal hyperplasia. Arch Sex Behav. (2004) 33:97–104. doi: 10.1023/B:ASEB.0000014324.25718.51
CrossRef Full Text | Google Scholar
78. Meyer-Bahlburg HFL, Dolezal C, Baker SW, New MI. Orientação Sexual em mulheres com hiperplasia adrenal congénita clássica ou não clássica em função do grau de excesso androgénico pré-natal. Arch Sex Behav. (2008) 37:85–99. doi: 10.1007 / s10508-007-9265-1
PubMed Abstract | CrossRef Full Text | Google Scholar