a esclera Azul é causada pela espessura e transparência das fibras de colágeno da esclera que permitem a visualização da uvea subjacente (Figura 1). A esclera é a camada exterior branca do olho, circundando a íris pode ser diluída em doenças congênitas, tais como osteogênese imperfeita 1). Outras doenças associadas com a esclera azul inclui vários dos tecidos conjuntivos, tais como frágil córnea síndrome de 2), Marshall e Defensor síndrome 3), POEMS (polineuropatia, organomegalia, endocrinopatia, proteína monoclonal e alterações da pele), síndrome 4), síndrome de Marfan, de Ehlers–Danlos, pseudoxanthoma elasticum, e Willems De Vries síndrome, para citar alguns. Doenças ósseas e do sangue também estão na lista incluem anemia Diamond-Blackfan, anemia grave deficiência de ferro, doença de Paget juvenil, e deficiência de ácido fosfatase 5).formas graves de Osteogénese Imperfeita são mais frequentemente diagnosticadas no início da vida, mas casos ligeiros podem não ser notados até mais tarde na vida. A cor Azul-cinza da sclera é devido às veias coroidais subjacentes que mostram através. Isto deve-se ao facto de a esclera ser mais fina do que o normal, uma vez que o colagénio de tipo I defeituoso não se está a formar correctamente 6). A sclera é uma estrutura densa de tecido conjuntivo mal vascularizado composta por colágeno dos tipos I, III, IV, V, VI e VIII, bem como elastina, proteoglicanos e glicoproteínas 7).nos Estados Unidos, a incidência de osteogênese imperfeita é estimada em um por 20 mil nascimentos vivos. Estima-se que 20.000 a 50.000 pessoas são afetadas pela osteogênese imperfeita nos Estados Unidos 8).as pessoas com Osteogénese Imperfeita nascem com tecido conjuntivo defeituoso ou sem capacidade para o fazer, geralmente devido a uma deficiência de colagénio tipo I. Esta deficiência surge de uma substituição aminoácida da glicina para aminoácidos mais volumosos na estrutura do colagénio tripla hélice. Como resultado, o corpo pode responder hidrolisando a estrutura imprópria do colágeno 9). Se o corpo não destruir o colágeno impróprio, a relação entre as fibrilhas de colágeno e cristais de hidroxiapatita para formar o osso é alterada, causando fragilidade. Como um distúrbio genético, osteogênese imperfeita tem sido historicamente visto como um transtorno autossômico dominante do colágeno tipo I. A maioria dos casos foi causada por mutações nos genes COL1A1 e COL1A2 10). Nos últimos anos, tem havido a identificação de formas autossômicas recessivas. Pelo menos sete subconjuntos foram descritos, embora quatro subtipos principais sejam mais comuns e variam de leve a grave. Indivíduos com osteogênese tipo I imperfeita têm pouca deformidade óssea, esclera azul persistente, perto da altura normal por idade adulta, e um>50% de chance de perda auditiva por idade adulta. Os doentes com Osteogénese Imperfeita perinatal letal (Tipo II) apresentam a maior gravidade, com múltiplas fracturas no útero ou no parto. Estes pacientes normalmente nascem mortos ou morrem cedo. A gravidade da Osteogénese Imperfeita depende do defeito genético específico. A maioria dos casos de osteogênese imperfeita são herdados de um pai. No entanto, alguns casos são o resultado de novas mutações genéticas. Uma pessoa com Osteogénese Imperfeita tem 50% de probabilidade de transmitir o gene e a doença aos seus filhos 11).
Figura 1. Anatomia ocular
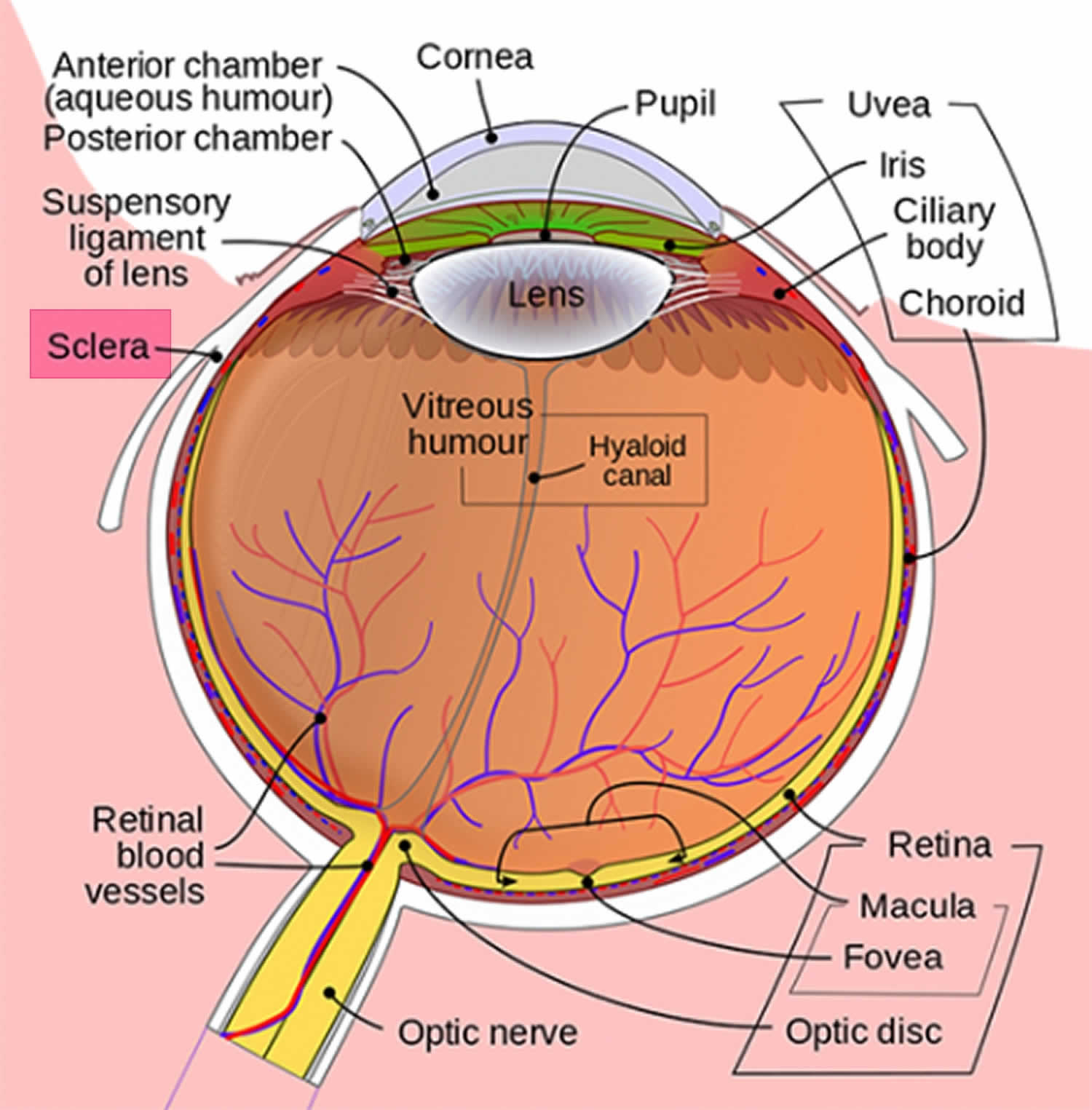
Figura 2. Azul esclera
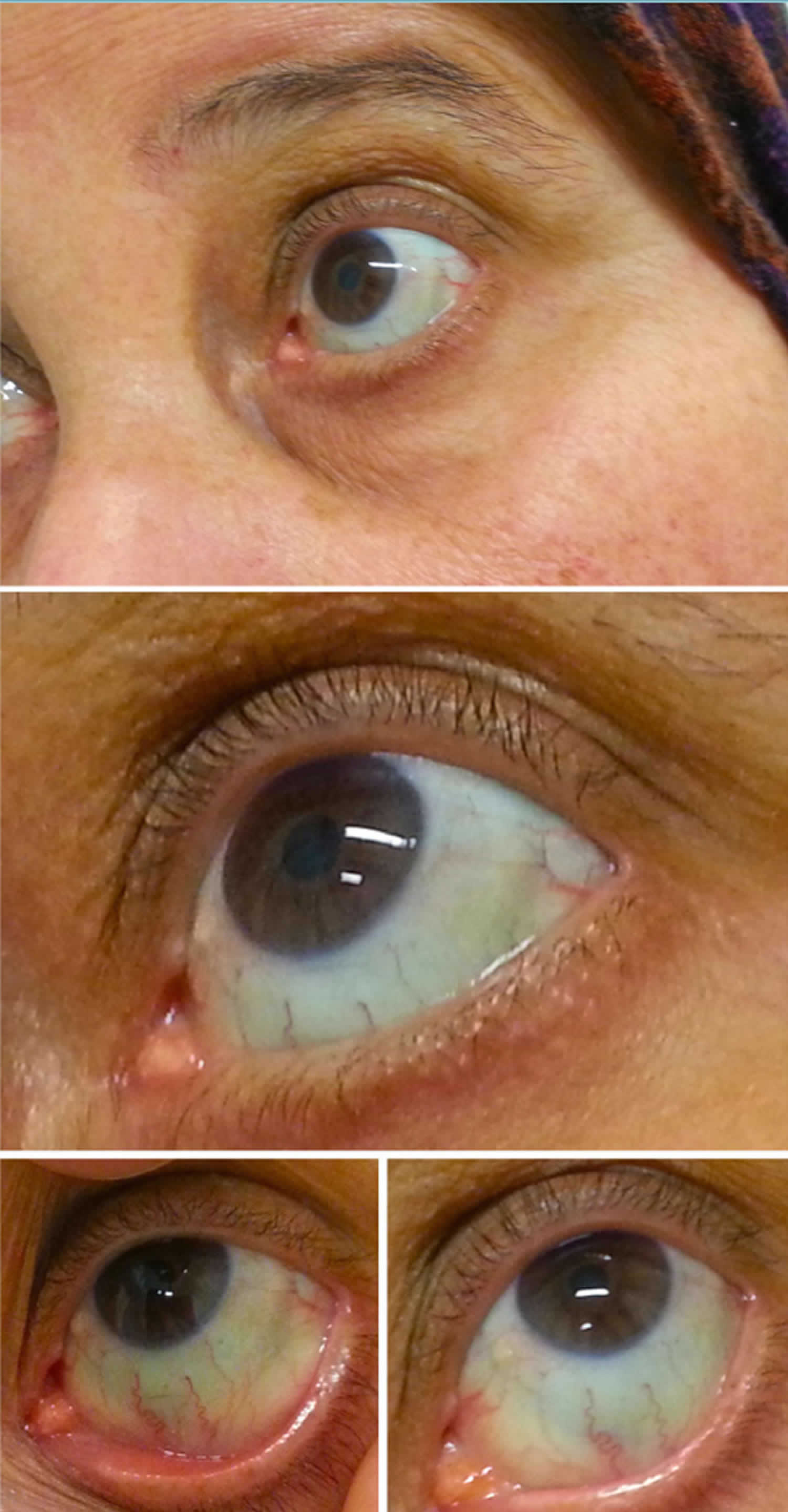
esclera Azul faz com
Azul esclera é a mais consistente manifestação na osteogênese imperfeita, que resulta de uma mutação no COLIA1 e COL1A2, para a codificação de tipo I procolágeno. Contudo, as classificações desta condição (tipos IV–VI) foram identificadas com esclerosantes normais. A Osteogénese Imperfeita está também associada à fragilidade anormal dos ossos e à surdez.
córnea quebradiça, esclera azul e cabelo ruivo estão associados com a síndrome da córnea quebradiça, uma condição que também apresenta anomalias esqueléticas,dentais e cutâneas 13). Uma mutação missense no ZNF469 foi encontrada como causadora de doença.
Outras condições subjacentes associados com a formação de esclera azul incluem Ehlers-Danlos (tipo VI), pseudoxanthoma elasticum, córnea plana, periféricos sclerocornea, buphthalmos, ceratocone, keratoglobus, alta miopia, ciliar/equatorial staphyloma, oculodermal melanocytosis e microcornea. Raramente, a sclera azul ocorre com síndrome de Hallermann-Streiff, síndrome de Marfan, síndrome de Turner, síndrome de Cheney, síndrome de Menkes, pyknodysostose, córneas quebradiças, ou displasia ectodérmica.a esclera azul pode também ocorrer em lactentes normais durante os primeiros meses de vida; no entanto, a persistência da descoloração azul ao longo do tempo pode sugerir a presença de pressão intra-ocular elevada. Os recém-nascidos prematuros frequentemente demonstram esclerosos azuis, particularmente os de origem caucasiana.
sclera Azul também pode ocorrer em isolamento como uma anomalia autossómica dominante ou autossómica recessiva hereditária 14).
os sintomas da esclera azul
a esclera Azul apresenta uma aparência azulada à sclera e pode estar associada a causa patológica ou não patológica. Outras características oculares das afecções dos tecidos conjuntivos associadas à esclera azul incluem córnea fina, pregas epicantais, miopia, Queratocone e estrias angióides. As características sistémicas das afecções dos tecidos conjuntivos associadas à esclera azul incluem anomalias cutâneas, anomalias cardíacas, cifoscoliose, Hipermobilidade articular, ossos frágeis, anomalias auditivas, anomalias vasculares e anomalias gastrintestinais 15).diagnóstico da esclera azul avaliação de diagnóstico da esclera azul exame externo, biomicroscopia da lâmpada de fenda e avaliação sistémica de distúrbios associados.Embora não exista um teste definitivo para a Osteogénese Imperfeita, a análise genética pode confirmar ou excluir mutações conhecidas 16).o tratamento com esclera azul para a esclera azul envolve diagnóstico e tratamento da causa subjacente. A esclera azul é principalmente devido a síndromes genéticas e, em menor extensão, em distúrbios nongenéticos e pode ocorrer como um efeito colateral da ingestão de medicação. A esclerose azul é comumente associada a doenças congênitas de síntese de colágeno, tais como osteogênese imperfeita, síndrome de Marfan, síndrome de Ehlers–Danlos, pseudoxanthoma elasticum, e síndrome de Willems De Vries, para citar alguns. Doenças ósseas e do sangue também estão na lista incluem anemia Diamond–Blackfan, anemia grave deficiência de ferro, doença Juvenil Paget, e deficiência de ácido fosfatase 17). Esclerosas azuis adquiridas foram descritas em adultos com melanose ocular, melanoma conjuntival, alkaptonúria e doença de Addison. Podem ser indicados referenciais apropriados para avaliações pediátricas e ortopédicas para manifestações não oculares 18).o aconselhamento genético para dissidentes hereditários Associados pode ser benéfico para doentes com esclerose azul e outras manifestações de doença sistémica.a intervenção cirúrgica pode ser indicada em casos de desbaste e Perfuração extremas. O suporte estrutural pode ser fornecido por uma fáscia lata orautóloga do enxerto escleral preservada, particularmente em casos que requerem sutura de um dispositivo como um implante de tubo 19). As investigações sistémicas e o tratamento devem ser considerados para abordar a patologia subjacente.
prognóstico da esclera azul
prognóstico varia com a presença de manifestações oculares e sistêmicas da doença subjacente. Os doentes com esclera Azul apresentam um risco aumentado de ruptura do globo ou perfuração escleral intra-operatória durante a cirurgia ocular de rotina. As afecções sistémicas são também prevalentes em doentes como a fístula carótida-cavernosa, ruptura arterial, perda auditiva e fracturas ósseas 20).
Referências