- Definitions and Prevalence
- diagnoza
- objawy NCCAH u kobiet
- objawy w dzieciństwie
- objawy w okresie dojrzewania i dorosłego życia
- leczenie NCCAH od pierwszej manifestacji do dorosłego życia
- Postępowanie w dzieciństwie
- Postępowanie w okresie dojrzewania
- pacjenci leczeni od dzieciństwa
- kobiety z pierwszym objawem w okresie dojrzewania lub objawami Hiperandrogennymi po przerwaniu leczenia
- NCCAH i reprodukcja
- Subfertility
- poronienia
- planowanie płodności
- prenatalne leczenie CAH
- leczenie w czasie ciąży
- inne szczególne problemy
- Zarządzanie stresem w NCCAH
- aspekty Psychobiologiczne w NCCAH
- wnioski
- wkład autora
- Oświadczenie o konflikcie interesów
Definitions and Prevalence
Congenital adrenal hyperplasia (Cah) includes a family of autosomal recesywny disorders characteristic by slightly to acutely defected kortyzol synthesis due to a deficient in one of the five adrenal steroidogenic enzymes required for cortisol production (1, 2). Konwencjonalnie, CAH dzieli się na (a) klasyczny (CCAH), przedstawiający z marnowaniem soli lub prostą formą wirylizującą, która manifestuje się przy urodzeniu i/lub w okresie noworodkowym, oraz (b) nieklasyczny (NCCAH), reprezentujący mniej poważną formę zaburzenia, które nie ma wieloznaczności narządów płciowych, nie jest natychmiast zagrażające życiu i prezentuje się w późniejszym życiu lub pozostaje bezobjawowa, lub jest błędnie diagnozowana jako inna choroba (3). Jednak granice między różnymi formami CAH nie są ściśle określone, co ma tendencję do zwiększania wyzwań związanych z tym zaburzeniem. Dlatego wskazane jest rozważenie CAH jako kontynuacji fenotypów, od ciężkich do łagodnych lub bezobjawowych (3).
najczęstszą formą CAH jest niedobór 21-hydroksylazy (21OHD), co powoduje upośledzenie lub brak konwersji 17 hydroksyprogesteronu (17 OHP) do 11 deoksykortyzolu, a progesteronu do deoksykortykosteronu. Blokada konwersji steroidów powoduje zwiększoną produkcję prekursorów androgenów, pod stymulacją CRH-ACTH, co prowadzi do biochemicznego hiperandrogenizmu, naznaczonego podwyższonym poziomem 17-OHP. Klasyczna postać choroby, występuje w 1 na 16 000 żywych urodzeń na całym świecie. Należy jednak wspomnieć, że postrzegana częstość występowania choroby jest związana z zastosowaną metodą przesiewową. Na przykład odnotowano, że częstość występowania klasycznej postaci CAH w Szwecji wynosi 1:11 500, gdy stosuje się podejście do badania przypadku, a liczba ta spada do 1:9 800, gdy stosuje się badania hormonalne. Podobnie, częstość występowania we Francji i Szwajcarii waha się między 1:15 472 i 1:23 000 oraz 1:10 749 i 1:11 661, gdy stosowane są różne metody badań przesiewowych (4).
NCCAH jest znacznie częstsza, występuje u około 1 na 1000 osób rasy kaukaskiej i częściej w niektórych grupach etnicznych, takich jak Żydzi aszkenazyjscy (1:27), Hiszpanie (1:53), Jugosłowianie (1:62) i Włosi (1:300) (5, 6). Niemniej jednak brakuje danych na temat częstości występowania NCCAH w oparciu o różne metody szacowania (badanie przypadku, badania hormonalne i badania molekularne). NCCAH jest uważane za najczęstsze autosomalne recesywne zaburzenie endokrynologiczne o częstotliwości nośnej od 1:25 do 1: 10. W NCCAH ze względu na 21-OHD rezydualną aktywność enzymatyczną szacuje się na około 10-70% (7-9).
diagnoza
w porównaniu z diagnozą klasycznej postaci choroby, która jest dokonywana przy urodzeniu lub w okresie noworodkowym z powodu wieloznaczności narządów płciowych i/lub objawów marnowania soli lub poprzez programy przesiewowe stosowane w niektórych krajach większość przypadków NCCAH nie jest łatwo wykrywalna (4, 10). Ponadto wiele osób pozostaje bezobjawowych w dzieciństwie i okresie dojrzewania, ma normalne funkcje rozrodcze i dowiaduje się o NCCAH tylko z powodu diagnozy innego członka rodziny i wynikających z tego badań (11). Jednak większość kobiet z NCCAH szukać pomocy medycznej, gdy występują objawy nadmiaru androgenów i, gdy podejrzenie kliniczne skłania do badania, podwyższone podstawowe 17 OHP poziomy będzie bardziej prawdopodobne niż nie wskazują na diagnozę NCCAH.
rzeczywiście, wytyczne kliniczne zaproponowane przez Towarzystwo endokrynologiczne zalecają podstawową, Nie stymulowaną wartość 17 OHP jako narzędzie przesiewowe dla NCCAH. Poranne poziomy 17 OHP >6 nmol/L w fazie pęcherzykowej u kobiet miesiączkujących powinny skłonić do dalszej oceny, ponieważ wykazano, że poziomy powyżej 6 nmol/L wychwytują 90% osób z NCCAH (12, 13). Losowe pomiary 17 OHP nie okazały się pomocne, ponieważ często dają one prawidłowy poziom u pacjentów z NCCAH, a ponadto są niezwykle wysokie w fazie lutealnej u połowy zdrowych kobiet (13). Jednak w naszych danych pochodzących od 280 osób z chorobą, sześciu pacjentów (2.1%) miało wyjściową wartość 17 OHP < 6 nmol/L (14). Dodatkowo Bidet i in., w dużej kohorcie kobiet z NCCAH zweryfikowanych technikami molekularnymi stwierdzono również podstawowe wartości 17 OHP niższe niż 6 nmol/L u 8% badanych (15). Wreszcie, na podstawie danych zebranych przez Speiser et al., 9% osób z NCCAH wykazywało wartości 17 OHP niższe niż 2 ng / ml (co odpowiada 6nmol/L). Według innych badań wartość wyjściowa 17 OHP pomiędzy 5,1 a 9 nmol / L jest wystarczająca do rozpoznania NCCAH (13, 16, 17). Ostatnio poziom podstawowy 17 OHP 4.6 nmol / L zaproponowano jako próg dla testów ACTH do przewidywania NCCAH u pacjentów z przedwczesnym nadnerczem w dzieciństwie(18).
suma tych wyników i sugestii wskazuje, że wybór pacjentów, którzy zostaną poddani testowi stymulacji Synakthenowej, powinien być oceniany w każdym przypadku. Do diagnozy wymagany jest poziom 17 OHP po stymulacji powyżej 3 nmol/L (19). Heterozygota posiadająca jedną mutację CYP21A2 wykazuje nieznacznie podwyższony poziom 17 OHP po stymulacji ACTH, chociaż występuje nakładanie się u osób nienaruszonych (9). Jednak, jak Dacou-Voutetakis et al. zwrócili uwagę, że jeśli suma wartości podstawowych i po stymulacji 17 OHP przekracza 1,5 nmol/L, to możliwość heterozygotyzmu jest wyjątkowo wysoka (20).
kolejna ważna uwaga dotyczy technik stosowanych do oceny 17 OHP. Poziomy 17 OHP są mierzone za pomocą różnych metod testów immunologicznych, ale jak ostatnio wykazano, najbardziej dokładne i wiarygodne wyniki uzyskano dzięki zastosowaniu kombinacji chromatografii cieczowej ze spektrometrią mas (LC-MS / MS). Rzeczywiście, wiele fałszywie dodatnich pomiarów 17 OHP stwierdzono, gdy pomiary LC-MS/Ms porównano ze standardowymi metodami (21) niemniej jednak te ostatnie procedury nie są powszechnie stosowane, w przypadku niepewnej diagnozy dostarczą bardziej precyzyjnych wyników. Należy zauważyć, że progi badań przesiewowych i diagnostycznych dla LC-MS/MS oznaczonych ilościowo 17 OHP nie zostały jeszcze określone. Progi te prawdopodobnie będą niższe niż te ustalone za pomocą testów immunologicznych ze względu na zwiększoną swoistość LC-MS/MS, metody mniej podatnej na reaktywność krzyżową i zakłócenia (22, 23). Należy wyjaśnić, czy profil sterydów w moczu jest wymagany do ostatecznej diagnozy (22). W przypadkach granicznych wskazane jest uzyskanie pełnego profilu kory nadnerczy po teście stymulacji ACTH w celu odróżnienia niedoboru 21-hydroksylazy od innych defektów enzymów i ustalenia jednoznacznej diagnozy.
w szczególności należy wykonać pełny profil sterydów w dwuznacznych przypadkach, aby potwierdzić niedobór 21-hydroksylazy i wykluczyć inne wykrywanie enzymatyczne. Włączenie 17 OHP, kortyzolu,11-deoksykortykosteronu,11-deoksykortyzolu, 17-OH-pregnenolonu, dehydroepiandrosteronu i androstendionu do profilu steroidowego w surowicy byłoby przydatne w celu wykluczenia innych przyczyn CAH, takich jak niedobór 11β-hydroksylazy i, rzadziej, niedobór dehydrogenazy 3β-hydroksysteroidowej lub niedobór oksydoreduktazy P450 (24). Chociaż badania genetyczne nie są obecnie uważane za podstawowe narzędzie diagnostyczne dla NCCAH, jest to obowiązkowe dla potwierdzenia diagnozy i poradnictwa genetycznego (3).
jeśli chodzi o poziom kortyzolu, ogólnie oczekuje się, że wartość po stymulacji wyniesie co najmniej 496 nmol/l. Na uwagę, według Stoupa et al., 60% z 47 dzieci z NCCAH w wyniku 21 OHD miało niskie wartości kortyzolu po stymulacji, co wskazuje na potrzebę zwiększonego nadzoru nad rozwojem niewydolności nadnerczy podczas dużych zdarzeń stresowych (25).
objawy NCCAH u kobiet
kliniczna ekspresja NCCAH charakteryzuje się wysokim poziomem polimorfizmu nie tylko w odniesieniu do wieku wystąpienia, ale także różnych objawów przedmiotowych i podmiotowych. Podaje się, że pierwsza kliniczna prezentacja NCCAH występuje w 11% przypadków przed ukończeniem 10 lat i w 80% w wieku od 10 do 40 lat (12). Korelacja genotypowo-fenotypowa w CAH i NCCAH nie została jeszcze wyjaśniona. Speiser i in. sugerują, że większość, ale nie cała zmienność fenotypowa niedoboru 21-hydroksylazy wynika ze zmienności allelicznej CYP21A2 (26).
objawy w dzieciństwie
w okresie noworodkowym kobiety zwykle pozostają bezobjawowe i mają normalne zewnętrzne narządy płciowe. Najwcześniejszym zgłoszonym przypadkiem NCCAH jest 6-miesięczna dziewczynka, u której rozwinęły się włosy łonowe (11). Zwykle wyniki kliniczne i objawy w przypadkach NCCAH zaczynają się od wieku 5 lat lub nawet później i są związane ze zwiększonym poziomem androgenów, chociaż w niektórych przypadkach może również wystąpić łagodny niedobór kortyzolu (8). W 92% przypadków NCCAH pierwszym objawem, który objawia się jest przedwczesny pubarche (PP), który występuje w wieku poniżej 8 lat u dziewcząt i poniżej 9 lat u chłopców (12) z szacunkową częstością występowania między 10 a 11,3% (15). Należy zauważyć, że u dzieci, o których mowa z PP, według różnych badań, częstość występowania NCCAH waha się od 5 do 30% (20, 27). Jednak na podstawie naszej analizy 280 pacjentów z molekularnym potwierdzeniem NCCAH stwierdziliśmy, że częstość występowania PP u 94 kobiet w wieku poniżej 8 lat wynosiła aż 88%. Z drugiej strony, analiza 45 mężczyzn z NCCAH wykazała PP tylko u 29% badanych (14). Ponadto należy pamiętać, że około połowa osób z PP może być heterozygotowymi nosicielami mutacji CYP21A2.
kolejnym aspektem, który wymaga wyjaśnienia, jest związek między PP a przedwczesnym dojrzewaniem. Istnieje hipoteza, że w niektórych przypadkach obwodowe aromatyzowanie androgenów nadnerczy do estrogenów może aktywować oś podwzgórze-przysadka-gonada, prowadząc do wtórnego przedwczesnego dojrzewania (28). W takim przypadku konieczne jest regularne monitorowanie. Niektóre dzieci rozwijają hirsutyzm, trądzik i / lub szybki wzrost oraz manifestują wysoką strukturę i zaawansowanie wieku kostnego (29). Te ostatnie mogą powodować ściętą końcową wysokość w wyniku szybkiego połączenia nasadowego. Inne rzadkie objawy kliniczne w dzieciństwie obejmują przyczepność wargową, owłosienie odbytu, łechtaczkę i chrypkę głosu (30-32).
objawy w okresie dojrzewania i dorosłego życia
w okresie dojrzewania i dorosłości NCCAH u kobiet wykazuje objawy hiperandrogenne przypominające zespół policystycznych jajników (PCOS) (33). Objawy obejmują hirsutyzm, trądzik, łysienie androgenowe, brak owulacji, zaburzenia miesiączkowania i niepłodność. W wieloośrodkowym badaniu najczęstszymi objawami wśród młodzieży i dorosłych kobiet były hirsutyzm (59%), oligomenorrhea (54%) i trądzik (33%). Wśród 161 kobiet z NCCAH objawami były hirsutyzm (78%), zaburzenia miesiączkowania (54,7%) i zmniejszenie płodności (12%) (34). W naszej grupie 161 kobiet z chorobą 63% pacjentek miało fenotyp podobny do policystycznego jajnika (14). To odkrycie ma znaczenie odwrotne, ponieważ częstość występowania NCCAH jest wysoka u kobiet z hiperandrogenemią. Oczywiście, badanie przeprowadzone przez Witcel et al. zgłaszanie częstości występowania NCCAH w zakresie od 1 do 33% u kobiet hiperandrogennych jest bardzo interesujące. W innym badaniu z udziałem 950 kobiet, które zostały skierowane do oceny nadmiaru androgenów, NCCAH wykryto u 4,2% z nich. Jednak pacjenci z NCCAH byli młodsi i bardziej hirsute w porównaniu z innymi grupami pacjentów hiperandrogennych. Charakteryzowały się również znacznie wyższym poziomem testosteronu, wolnego testosteronu i 17 OHP niż pacjenci z innymi zespołami hiperandrogennymi. W badaniu ultrasonograficznym jajników u 77% z nich stwierdzono policystyczne jajniki, a u 41% zwiększono rozmiar jajników. Podsumowując, spełniły one kryteria PCOS zgodnie z NIH i Rotterdamem w wysokości odpowiednio 56% i 72,8% (35).
postępujący charakter choroby podkreśla fakt, że częstość występowania hirsutyzmu zwiększa się wraz z wiekiem i zaobserwowano rzadkie występowanie przed okresem dojrzewania. Hirsutyzm jest najczęstszym objawem klinicznym zgłaszanym u pacjentów z NCCAH, wynoszącym od 71 do 96% (28, 36). Dziewczęta w okresie dojrzewania z NCCAH zazwyczaj występują z hirsutyzmem (37, 38). Na drugim końcu spektrum jest problem łysienia. Łysienie typu męskiego jest zgłaszane u pacjentów z NCCAH. Częstość występowania łysienia również rosła wraz z wiekiem, z 6% u pacjentów w drugiej dekadzie życia do 19% w piątej dekadzie życia, co ponownie wskazuje na postępujący charakter choroby (39). Trądzik występuje u prawie 33% pacjentów z NCCAH (36). Co ciekawe, mutacje genu CYP21A2 powodującego NCCAH wykryto u 4,9% ze 123 dorosłych kobiet z ciężkim trądzikiem (40).
nieprawidłowości miesiączkowania, w tym oligomenorrhea lub wtórny brak miesiączki, często mogą być oznaką NCCAH u osób post menarche. Dodatkowo, 8-9% przypadków również wystąpić pierwotny brak miesiączki i, z tego powodu, zasięgnąć porady lekarza po raz pierwszy. Ponadto wśród osób NCCAH oligomenorrhea jest bardziej powszechne niż pierwotny brak miesiączki. Na przykład w serii z Moran et al., wśród kobiet z NCCAH w wieku od 10 do 19 lat, oligomenorrhea była obecna w 56% przypadków w porównaniu do tylko 9% Okresu Dojrzewania z pierwotnym brakiem miesiączki (12). Podsumowując, tak wiele objawów NCCAH przypominają te PCOS, że nie jest zaskakujące, że NCCAH został nazwany Wielkim Naśladowcą PCOS. W każdym przypadku rozpoznanie NCCAH należy rozważyć podczas oceny każdej młodej kobiety, która jest kierowana do objawów hiperandrogennych.
w badaniu przeprowadzonym przez Arlt et al., pacjenci z NCCAH charakteryzowali się istotnie wyższym BMI i niższym końcowym wzrostem w porównaniu do dopasowanych grup kontrolnych (41). Dane te, w połączeniu z kilkoma badaniami wskazującymi na obecność niewrażliwości na insulinę, sugerują niekorzystny profil metaboliczny, który może być patofizjologicznie wyjaśniony przez wpływ podwyższonego stężenia androgenów i / lub w wyniku leczenia glukokortykoidami (42, 43). Osteoporozę można również wykryć u tych pacjentów, prawdopodobnie jako konsekwencję leczenia kortykosteroidami (34).
leczenie NCCAH od pierwszej manifestacji do dorosłego życia
Po ustaleniu diagnozy NCCAH należy rozważyć kilka kwestii związanych z późniejszymi zaleceniami leczenia. Pierwszym pytaniem, które należy rozwiązać, jest to, czy wskazane jest leczenie glikokortykosteroidami (GC). Ogólne rozumowanie tego podejścia polega na tym, że poprzez zapewnienie pacjentowi wystarczających stężeń kortyzolu, jego codzienne potrzeby są pokrywane, aw konsekwencji stymulacja osi CRH-ACTH będzie zwężana, co prowadzi do zmniejszenia produkcji androgenów nadnerczy. Ogólna zasada dotycząca tego podejścia polega na tym, że GCs podaje się w dawkach zastępczych, a nie farmakologicznych, podczas gdy wpływ wieku, płci, danych laboratoryjnych, zaleceń specyficznych dla pacjenta i celów leczenia na glikokortykosteroidową terapię zastępczą jest poważnie brany pod uwagę. Jednakże, powinniśmy również mieć świadomość, że długotrwałe leczenie substytucyjne GC może prowadzić do hiperkortyzolemii, z wynikającym dobrze udokumentowanym niekorzystnym wpływem na każdy aspekt metabolizmu, zwłaszcza wzrost, rozkład tłuszczu i insulinooporność, a także na profil psychologiczny. Inną poważną wadą tego podejścia jest brak odpowiedniego wskaźnika klinicznego lub biochemicznego markera odpowiedniej dawki zastępczej, takiego jak istnieją w odniesieniu do wartości TSH w niedoczynności tarczycy.
wreszcie fakt, że nie ma konsensusu co do tego, które GC powinno być stosowane i brak długoterminowych danych dotyczących różnych sposobów podawania GC, dodatkowo komplikuje decyzje lekarzy prowadzących i wybór optymalnego wyboru dla każdego pacjenta. Hydrokortyzon jest zwykle stosowany u dzieci, ponieważ najbardziej przypomina naturalny hormon (kortyzol), ale nie uważa się go za odpowiednie podejście u młodzieży i młodych kobiet ze względu na potrzebę wielokrotnego codziennego dawkowania. Dlatego większość dorosłych endokrynologów preferuje deksametazon lub prednizolon w odpowiednich dawkach. Z drugiej strony należy podkreślić, że równoważność różnych GCs opiera się na ich działaniu przeciwzapalnym, a nie na różnych aspektach ludzkiego metabolizmu. Ponieważ kortyzol wpływa na prawie 20% ludzkiego genomu, oczekuje się różnych odpowiedzi różnych GCs w różnych tkankach. Potwierdzeniem tej percepcji jest fakt, że podawanie deksametazonu pacjentom z CAH wykazało pogorszenie wskaźników insulinooporności w porównaniu z innymi GCs (44). Ponadto podawanie 2,5-7,5 mg prednizolonu, dawki uważanej za normalną, wywiera długotrwały negatywny wpływ na metabolizm kości (45). Dlatego hydrokortyzon należy uznać za najlepszą formę leczenia w przypadku suplementacji GC. Pojawienie się nowo zsyntetyzowanej formuły hydrokortyzonu z jedną tabletką dziennie i jej początkowe pozytywne wyniki u pacjentów z CAH pokazują wiele obiecujących na przyszłość (46).
Postępowanie w dzieciństwie
pacjenci z NCCAH, u których w dzieciństwie rozpoznano objawy PP, mogą być leczeni hydrokortyzonem w celu zahamowania hormonów nadnerczy i zapobiegania szybkiemu postępowi wieku kostnego, który może mieć wpływ na ostateczny wzrost. U pacjentów, u których leczenie hydrokortyzonem rozpoczęto na 1 rok przed rozpoczęciem okresu dojrzewania i u których wiek kostny wynosił poniżej 9 lat, ostateczny wzrost pozostawał w granicach potencjału genetycznego (47). Dostępne badania wskazują, że wzrost dorosłych osób zbliżał się do oczekiwanego wzrostu docelowego u pacjentów, którzy byli ściśle monitorowani i ściśle przestrzegali planów leczenia (48-50). Niedawne małe badanie pięciu przypadków (pacjenci w wieku 6,1–9,2 lat) wykazało, że ultralekka dawka deksametazonu jest obiecującą opcją zmniejszenia stresu endogennego i jego skutków. Czy stanowi to realistyczne podejście i ile podanej dawki powinno pozostać do wyjaśnienia (51). Badanie przeprowadzone przez Nebesio et al. porównano działanie hormonalne i farmakokinetykę hydrokortyzonu, prednizolonu i deksametazonu u 9 pacjentów przed okresem dojrzewania z CCAH. Wykazano, że deksametazon był silniejszy niż inne formy w osiąganiu znacznie niższych poziomów hormonów nadnerczy, co sugeruje, że deksametazon jest bardziej skuteczny w hamowaniu produkcji androgenów nadnerczy (46). Oczywiście potrzebne są dalsze badania aby zweryfikować lub odrzucić to ustalenie. Ponadto, w niektórych przypadkach podwyższone stężenia androgenów mogą prowadzić do wtórnej stymulacji osi GnRH, co prowadzi do przedwczesnego dojrzewania. W takim przypadku można przepisać przebieg równoległy z analogiem GnRH, jeśli wiek kostny jest znacznie wyższy niż wiek chronologiczny i/lub przewidywany ostateczny wzrost jest nieproporcjonalny do docelowego wzrostu.
kolejna ważna uwaga dotyczy przypadków współistniejącego niedoboru GH w przypadku, gdy leczenie analogami GnRH i receptą GH prowadziło do osiągnięcia docelowego wzrostu (52). Zgodnie z aktualnymi wytycznymi, wyżej opisana terapia jest ostrożnie zalecana tylko dla tych przypadków, w których przewidywany wzrost SD wynosi -2,25 < docelowy wzrost lub nawet niższy (53). Alternatywnym wskazaniem do rozpoczęcia leczenia hydrokortyzonem jest niewystarczająca odpowiedź kortyzolu po stymulacji ACTH (36).
preferowanym leczeniem GC u dzieci jest zwykle hydrokortyzon w dawce 10-15 mg/m2 pc., podzielony na trzy dawki. Często skuteczne okazały się również niższe dawki, począwszy od 6 mg/m2 pc./dobę (34). Należy unikać przedawkowania, biorąc pod uwagę, że może to skutkować słabym wzrostem, a także cechami Cushingoidów. Regularny wzorzec wzrostu, wiek kostny zgodny z wiekiem chronologicznym i brak otyłości Centralnej mogą również służyć jako wskaźniki kliniczne dla odpowiedniego postępowania. Jeśli chodzi o profil biochemiczny / hormonalny, poziomy androstendionu i testosteronu w średnich i górnych zakresach dla wieku kostnego są uważane za lepsze markery odpowiedniej terapii zastępczej GC u dzieci z NCCAH. Jednak tłumione poziomy testosteronu stwierdzono u 10% pacjentów z NCCAH, podczas gdy kolejne 28% pacjentów miało zwiększone stężenie testosteronu, zjawisko to prawdopodobnie można przypisać zmienności hydrokortyzonu (54, 55). Wartości DHEAS są zmniejszane przy bardzo małych dawkach, podczas gdy tłumienie poziomu 17 OHP i progesteronu wymaga bardzo wysokich dawek GC. Należy zauważyć, że wartości 17 OHP są kluczowe dla diagnozy, ale nie są pomocne podczas obserwacji. Należy pamiętać, że pobieranie próbek krwi do oceny hormonalnej musi być przeprowadzane bez przerywania leczenia. Jednak ze względu na zakłopotanie choroby i jej wieloaspektowy charakter, nie ma konkretnych wytycznych dotyczących czasu zmian schematu lub przerwania leczenia glukokortykoidami u dzieci.
Postępowanie w okresie dojrzewania
pacjenci leczeni od dzieciństwa
do czasu ustalenia prawidłowego wzorca menstruacyjnego u dziewcząt z NCCAH zaleca się kontynuację GCs, która rozpoczęła się w dzieciństwie (56). Jednak młodzież często nie wykazuje wystarczającej zgodności z przewlekłym podawaniem leków i często pomija dawki. Dodatkowo, w okresie dojrzewania, okres półtrwania hydrokortyzonu spada o 50% w wyniku zwiększonego poziomu IGF-1, co zmniejsza aktywność 11ßOHSD, jak również ze względu na zwiększony klirens kortyzolu wynikający z amplifikacji szybkości filtracji kłębuszkowej (57). U młodej kobiety, 2 lata po menarche i jeśli zarejestrowano normalne cykle owulacyjne, wysoce zalecane jest skoncentrowane na pacjencie podejście do objawów hiperandrogennych, które mogą się pojawić.
Jeśli wystąpią ciężkie objawy hiperandrogenne, takie jak hirsutyzm, trądzik i/lub oligomenorrhea, rozważana będzie kontynuacja leczenia. Ważne jest, aby pamiętać o delikatnej wrażliwości młodego nastolatka, który zostanie poważnie uszkodzony przez „odstraszające” oznaki hiperandrogenizmu. Połączenie objawów hiperandrogennych, zaburzeń miesiączkowania i złej jakości życia jest dobrze udokumentowane i jest szczególnie wysokie u młodszych kobiet. Inną kwestią, którą należy wziąć pod uwagę, jest odpowiedź na stymulację rezerwy nadnerczy po stymulacji ACTH. Biorąc pod uwagę, że szczyt kortyzolu kobiet w okresie dojrzewania i dorosłych po stymulacji wynosi poniżej 496 nmol/L, w przypadkach stresu należy zastosować leczenie steroidowe (58). Z drugiej strony, jeśli u młodego pacjenta nie wystąpi żaden z powyższych objawów, leczenie GC może zostać przerwane, po czym zaleca się regularną obserwację.
kobiety z pierwszym objawem w okresie dojrzewania lub objawami Hiperandrogennymi po przerwaniu leczenia
w przypadku wystąpienia objawów hiperandrogennych zaleca się podejście zorientowane na pacjenta, koncentrujące się na głównych dolegliwościach pacjenta. Jest to o wiele lepsze niż ogólny uniwersalny sposób działania, ponieważ przy tym drugim podejściu młody pacjent będzie rozczarowany, co doprowadzi do przerwania leczenia, a w rezultacie do niekorzystnych skutków zdrowotnych w wieku dorosłym. W przypadkach z ciężkim hirsutyzmem przebieg od 6 do 12 miesięcy z doustnymi pigułkami antykoncepcyjnymi (OCPs) stanowi rozsądną procedurę, biorąc pod uwagę korzystny wpływ OCPs na produkcję wątroby SHBG i zmniejszenie uwalniania androgenów z jajnika. Oczekuje się poprawy klinicznej po co najmniej 2-3 miesiącach rozpoczynania OCPs, podczas gdy jednoczesne stosowanie progestagenu o minimalnych właściwościach androgennych jest wysoce zalecane. W przypadku niepowodzenia OCP w podejściu pierwszego rzutu do leczenia można dodać antyandrogeny (spironolakton, flutamid, bikalutamid, octan cyproteronu i finasteryd). Z naszego doświadczenia wynika, że podawanie bikalutamidu osiągnęło znaczną poprawę w przypadkach ciężkiego trądziku, ale podobnych wyników nie uzyskano w przypadkach ciężkiego hirsutyzmu. Kosmetyczne podejścia, takie jak aplikacja laserowa i depilatory, mogą być również sugerowane dla kobiet skarżących się na nadmierny lub niechciany wzrost włosów lub wypadanie włosów na skórze głowy (łysienie androgenowe) (32, 34, 57).
u pacjentów z niedostatecznym wydzielaniem kortyzolu po stymulacji lub jeśli OCPs i antyandrogeny nie mogą być tolerowane, zaleca się Kurs z GCs (57). Dane z New et al. wskazują, że nieregularne miesiączki i trądzik są odwracane w ciągu 3 miesięcy po rozpoczęciu terapii glikokortykosteroidami, podczas gdy hirsutyzm wymaga prawie 30 miesięcy (59). W przeciwieństwie do dzieciństwa, w okresie dojrzewania, często stosuje się dłużej działające steroidy i zaleca się Schematy 5 mg prednizolonu lub 0,25 mg deksametazonu (53). Jednak w rzeczywistym świecie dane kliniczne wykazały wiele różnych schematów stosowanych w zarządzaniu NCCAH (41).
podstawowym celem leczenia powinna być ulga pacjenta od objawów hiperandrogennych. Tak więc, wśród osób z NCCAH zdiagnozowano wyłącznie na podstawie wyników laboratoryjnych, cechy kliniczne powinny kierować zalecenia dotyczące zarządzania, ponieważ wyniki hormonalne i molekularne niekoniecznie przewidują nasilenie kliniczne. Zgodnie z wytycznymi Towarzystwa Endokrynologicznego pacjenci z NCCAH powinni mieć możliwość przerwania leczenia GC, gdy objawy ustąpią (60). Oczywiście, nie należy tracić tych pacjentów do obserwacji, podczas gdy leczenie należy wznowić w przypadku nawrotu objawów. Ponadto, w przypadku przerwania leczenia, pacjentów należy poinformować o możliwości wystąpienia niepłodności i zachęcić do zasięgnięcia porady lekarza, jeśli chcą zajść w ciążę (19). Należy zauważyć, że odpowiednie przeniesienie pacjenta z pediatrycznego do endokrynologa dorosłych powinno być przeprowadzone optymalnie po 1 roku zsynchronizowanego monitorowania (53, 61).
NCCAH i reprodukcja
Subfertility
Subfertility jest często opisywana w klasycznych formach CAH, co jest głównie spowodowane zaburzeniami miesiączkowania, przewlekłym brakiem owulacji i deformacjami anatomicznymi. Wskaźnik urodzeń oszacowano na 17% w porównaniu do 65% populacji kontrolnej (62). Tymczasem dane dotyczące płodności u kobiet z NCCAH zostały ostatnio szczegółowo ocenione, a szacowana częstość niepłodności wynosi 11% wśród kobiet z NCCAH, to znaczy stosunkowo łagodniejsza niż w CAH, a w wielu przypadkach jest łatwa do rozwiązania. Bidet i in. oceniono płodność u 190 kobiet cierpiących na NCCAH, z których 95 chciało zajść w ciążę. W tej populacji odnotowano 187 ciąż u 85 kobiet, co dało 141 porodów u 82 osób. Należy podkreślić, że 99 ciąż (52,9%) wystąpiło przed rozpoznaniem NCCAH, trzy z nich z zastosowaniem protokołów indukcji owulacji, reszta była spontaniczna, podczas gdy 88 ciąż miało miejsce po rozpoznaniu NCCAH. U zdecydowanej większości z nich ciąża rozwinęła się po rozpoczęciu terapii hydrokortyzonem, natomiast u 11 kobiet wystąpiła spontanicznie (63, 64). W innym raporcie 22 obserwowane NCCAH kobiet pragnących ciąży, 12 ciąż nastąpiło z prednizonu (65).
poronienia
, odsetek poronień wynosił odpowiednio 6,5% i 26,3% u pacjentów leczonych GCs i nieleczonych (64). Wynik ciąży może być nawet udany bez leczenia glukokortykoidami w przypadkach, w których NCCAH nie został jeszcze zdiagnozowany, jak również opisano w opisie przypadku przez Cuhaci i wsp. Niemniej jednak, ta sama para doświadczyła dwóch poronień i zgłosiła subfertility po tej pierwszej ciąży(66). Jeśli chodzi o możliwość ciąż przedwczesnych, jak ocenia Bidet et al., nie różni się znacząco od reszty populacji (15).
planowanie płodności
dla tych kobiet z objawowym hiperandrogenizmem lub ze zgłoszoną niepłodnością, ale które chcą zajść w ciążę, terapia GC jest wysoce zalecana. W tym okresie stosowanymi glikokortykosteroidami są prednizon lub hydrokortyzon. Najważniejszym działaniem, ponieważ będzie ono kierować kolejnymi krokami, są badania genetyczne przyszłych rodziców. Kobiety z NCCAH, którzy pragną zajść w ciążę, powinny być świadome ryzyka urodzenia dziecka z klasyczną postacią choroby. Wyniki ciąż wśród kobiet z NCCAH, a dokładniej częstość występowania niemowląt urodzonych z CCAH, szacuje się w ostatnich badaniach na 1,5-2,5%, przy NCCAH na około 15% (36, 64). Oczywiście częstość ta zależy od populacji referencyjnej, a większa częstość występuje w populacjach z wysokim wskaźnikiem małżeństw międzyludzkich (36). Przewidywane szanse rodziców na narodziny dziecka z CAH wynoszą 1 na 120 dla nieznanego genotypu Ojcowskiego, zero, jeśli ojciec nie ma istotnej mutacji, i 1 na 4, jeśli ma mutację heterozygotyczną (19). W rezultacie badania genetyczne partnerów tych kobiet są niezbędne do oceny ryzyka urodzenia dziecka z klasyczną formą CAH (64, 67).
w przypadku, gdy przyszła matka nosi dwie mutacje NC (Tabela 1), nie ma bezwzględnej potrzeby badań genetycznych u przyszłego ojca, ponieważ jeśli jest on heterozygotyczny dla mutacji CYP12A2, potomstwo jest zagrożone rozwojem nieklasycznej postaci choroby w przyszłości. Należy jednak zauważyć, że status nosiciela wynosi 2-15% w różnych populacjach, a połowa tych osób nosi ciężką mutację. Ponadto, ostateczne zalecenia dotyczące sytuacji, w których badania genetyczne nie są wymagane, są trudne ze względu na niedoskonałą korelację genotypowo-fenotypową, szczególnie w przypadku łagodniejszych mutacji . Na przykład mutacja P30L, najczęściej związana z NCCAH, występuje również u pacjentów z klasycznym CAH . Duże, wieloośrodkowe badanie Europejskie wykazało ostatnio, że tak było u 78% pacjentów . W tym badaniu łagodna mutacja V218L była związana z klasyczną, a nie NCCAH w 30% przypadków. Dlatego wszystkim pacjentom z NCCAH należy zaoferować poradnictwo genetyczne i ocenę molekularną partnerów reprodukcyjnych.
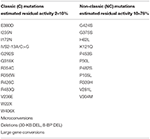
Tabela 1. Mutacje CYP21A2 o minimalnej (C) lub umiarkowanej (NC) aktywności enzymu resztkowego (14, 68, 69).
natomiast w przypadku, gdy przyszła matka jest heterozygotą złożoną z jedną ciężką (C) i jedną mutacją NC, wówczas obowiązkowe jest badanie genetyczne przyszłego ojca. Należy pamiętać, że dane pochodzące z badań z wykorzystaniem testów immunologicznych lub dokładniejszych procedur LC-MS / MS, konsekwentnie podkreślają niezdolność do niezawodnego wykluczenia heterozygotyzmu przy użyciu stymulowanych przez basal lub ACTH wartości 17 OHP, ze względu na znaczące nakładanie się z normalnym zakresem. Można zasugerować zastosowanie testu Synakthen, a jeśli nie jest on zgodny z heterozygotycznością (suma stymulowanych wartości podstawowych i szczytowych 17 OHP < 1,5 nmol / L), można uniknąć badania DNA. Powinniśmy jednak być bardzo ostrożni w interpretacji tego testu, ponieważ nie został on zatwierdzony w innych badaniach. Stąd zastosowanie stymulowanego ACTH 21-deoksykortyzolu pojedynczo lub jako część stosunku steroidowego lub profilu steroidowego może ułatwić biochemiczną identyfikację heterozygotów w przyszłości, szczególnie gdy LC-MS/MS staje się szerzej dostępny . Jeśli przyszły ojciec nosi mutację NC, nic więcej nie jest potrzebne. Odwrotnie, jeśli jest nosicielem klasycznej mutacji, pojawi się pytanie dotyczące prenatalnego leczenia płodu. Wszystkie możliwe kombinacje genotypów i sugerowane procedury przedstawiono w tabeli 2.

Tabela 2. Planowanie sugerowanych zabiegów w czasie ciąży na podstawie genotypu przyszłych rodziców.
prenatalne leczenie CAH
celem prenatalnego leczenia CAH jest zapobieganie wirylizacji narządów płciowych płodu, ale także złagodzenie stresu odczuwanego przez rodziców, którzy prawdopodobnie będą mieli dziecko z niejednoznacznymi genitaliami (70). Deksametazon jest stosowany ze względu na jego zdolność do przenikania przez łożysko i ponieważ nie jest dezaktywowany z dehydrogenazy steroidowej łożyska 11β OH I wiąże się tylko w minimalnym stopniu z globuliną wiążącą kortyzol we krwi matki (CBG). Deksametazon, hamując wydzielanie ACTH płodu, zmniejsza podwyższoną produkcję androgenów płodu. Leczenie należy przerwać w przypadku, gdy analiza kariotypu lub DNA ujawni odpowiednio samca lub nienaruszoną samicę (71). Niemniej jednak, mimo że wirylizacja narządów płciowych płodu rozpoczęła się już w 6-7 tygodniu po zapłodnieniu, biopsji kosmków kosmków kosmówkowych w diagnostyce genetycznej nie można uzyskać przed 10-12 tygodniem. Ten przedział czasowy sugeruje, że wszystkie ciąże zagrożone wirylizacją CAH powinny być leczone, mimo że dotyczy to tylko 1 z 8 płodów i kobiet (19, 34, 70).
to, czy ekspozycja płodu na Deksametazon w celach profilaktycznych jest akceptowalna z medycznego i etycznego punktu widzenia, pozostaje kontrowersyjne (70). Badania na zwierzętach wykazały, że Deksametazon w pierwszym trymestrze ciąży zmniejsza wagę urodzeniową, wpływa na czynność komórek beta trzustki i rozwój mózgu oraz predysponuje do lęku, nadciśnienia i hiperglikemii w wieku dorosłym (70). Ponadto 10-20% kobiet w ciąży stosujących leczenie deksametazonem w czasie ciąży skarży się na przyrost masy ciała, zwiększony apetyt, wahania nastroju, bezsenność, obrzęki, nadciśnienie, hiperglikemię i rozstępy. Jednak nowe et al. stwierdzono niższy niż średni wynik Pradera u płodów leczonych deksametazonem w okresie prenatalnym, ale brak różnicy w długoterminowych wynikach (72). Dodatkowo w recenzji Merce Fernandez-Balsells et al. wykazano, że deksametazon jest związany ze zmniejszeniem wirylizacji płodu bez znaczących działań niepożądanych u matki lub płodu (63). Autorzy wyżej wymienionej recenzji, jak również kolejne wytyczne Towarzystwa Endokrynologicznego, wskazują jednak, że ze względu na niewielkie rozmiary próbek w całej literaturze, temat pozostaje niepewny i konieczne jest dalsze badanie.
decyzja o rozpoczęciu leczenia powinna być podejmowana tylko w dużych ośrodkach z doświadczonym zespołem i protokołami zatwierdzonymi przez instytucjonalne Rady przeglądowe i w oparciu o wartości i preferencje rodziny oraz ich pisemną świadomą zgodę jako warunek wstępny (19, 63). Kilku specjalistów w tej dziedzinie wskazuje, że większą wartość należy położyć na zapobieganiu niepotrzebnej prenatalnej ekspozycji matki i płodu na deksametazon, zamiast nakładać emocjonalne żniwo niejednoznacznych narządów płciowych na rodziców i pacjentów. Co najważniejsze, należy wyjaśnić rodzicom, że podawanie deksametazonu nie zmienia stanu pacjenta, ale ma na celu zmniejszenie konieczności operacji, a nie zachowanie życia lub zdolności intelektualnych. Muszą również wiedzieć, że prawdopodobieństwo, że ich dziecko będzie cierpieć z powodu klasycznej postaci choroby, jest wysokie, pomimo leczenia. Co najważniejsze, narażenie młodego organizmu na bardzo silny GC w szczególnie wrażliwym okresie programowania i wzrostu płodu, co może okazać się bezużyteczne w przypadku płodu z kariotypem XY, nie jest obecnie poparte solidnymi i niepodważalnymi danymi.
najlepiej, aby była wczesna diagnoza, wykonywana za pomocą nieinwazyjnej techniki i przed rozpoczęciem wirylizacji narządów płciowych płodu. Pracując w tym kierunku, nowe badania wskazują na wykorzystanie wolnego od komórek DNA płodu uzyskanego z osocza matki jako obiecującej metody, która pozwoli na określenie płci płodu i rozpoznanie CAH we wczesnym wieku ciążowym (<9 tygodni). Jeśli procedura ta jest szeroko stosowana w praktyce klinicznej, uniknie się niepotrzebnego prenatalnego leczenia deksametazonem(73).
leczenie w czasie ciąży
w czasie ciąży kobiety z NCCAH powinny być leczone hydrokortyzonem, nie tylko dlatego, że ciąży zwykle towarzyszy podwyższony poziom stresu, ale także jako środek zapobiegawczy przeciwko wyżej wymienionej wysokiej częstości poronień. Ta forma glukokortykoidu jest preferowana ze względu na jej metabolizm przez enzym łożyskowy 11β OH dehydrogenaza steroidowa II, która zapobiega wpływowi glukokortykoidu na płód (71). W momencie poczęcia dawkę hydrokortyzonu należy zwiększyć do 20-25 mg / dobę, a modyfikacje dawki przeprowadza się co 6-8 tygodni w celu utrzymania poziomu testosteronu na górnym normalnym poziomie trymestru ciąży (74). Jako przykład przedstawiono wartości testosteronu i D4 u pacjenta z NCCAH od poczęcia do 3 lat po porodzie dwóch zdrowych bliźniąt (ryc. 1).
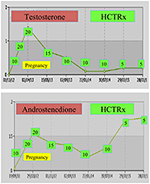
Rysunek 1. Poziomy testosteronu i andostenodionu oraz późniejsze dawkowanie hydrokortyzonu (HCTRx), od poczęcia do 3 lat po porodzie, podawane pacjentowi z NCCAH.
inne szczególne problemy
Zarządzanie stresem w NCCAH
podczas dużego zagrażającego życiu stresu, operacji lub ciężkiej choroby pacjenci z NCCAH leczeni glikokortykosteroidami mogą wymagać większych lub częstszych dawek glikokortykosteroidów, biorąc pod uwagę, że ich czynność nadnerczy jest tłumiona jatrogenicznie. Dlatego ważne jest, aby edukować rodziców małych dzieci, a także ponownie edukować pacjentów w zakresie przechodzenia do opieki nad dorosłymi, o dawkowaniu stresu. U pacjentów z NCCAH, którzy nie są leczeni GCs lub w przypadku przerwania leczenia, należy ocenić odpowiedź kortyzolu na ACTH. Prawie jedna trzecia pacjentów z NCCAH reaguje nieprawidłowo na stymulację (15, 75). Dla osób, które normalnie reagują na stymulację ACTH, nie zaleca się leczenia dawkami stresu (19). Leczenie mineralokortykoidami nie jest wymagane w żadnym z przypadków, ponieważ u tych pacjentów występuje normalne wydzielanie aldosteronu i nie dochodzi do zaniku soli (58). Należy zauważyć, że w przypadku nadczynności tarczycy lub zwiększenia leczenia lewotyroksyną zwiększa się klirens kortyzolu i może wystąpić przełom nadnerczy (58, 76).
aspekty Psychobiologiczne w NCCAH
Meyer-Bahlburg et al. czy w kilku badaniach odnotowano upośledzenie profilu psychologicznego u pacjentów z NCCAH z powodu niedoboru 21OH. Mówiąc dokładniej, dotknięte kobiety miały wyższy wynik maskulinizacji / defeminizacji na kilku miarach zachowań związanych z płcią w porównaniu z normalnymi kobietami kontrolnymi, chociaż znacznie mniej niż u kobiet z klasycznym CAH. Miały one również znacznie zwiększoną orientację biseksualną lub homoseksualną. Retrospektywne wywiady kliniczno-jakościowe z tymi kobietami ujawniły historię dyskomfortu i stresu społecznego związanego z ich doświadczeniami przed leczeniem z objawami zależnymi od androgenów, takimi jak trądzik, hirsutyzm i trudności z poczęciem (59, 77, 78). Podobnie Arlt et al. wykazały, że subiektywny stan zdrowia został znacznie osłabiony we wszystkich ośmiu domenach krótkoterminowego Badania Zdrowia, z najbardziej widocznymi różnicami w porównaniu z kontrolami dopasowanymi do wieku i płci, dotyczącymi ogólnego stanu zdrowia, witalności i ograniczeń roli z powodu problemów emocjonalnych. Kwestionariusz szpitalnej skali lęku i depresji (ang. Hospital Anxiety and Depression Scale, HADS) również wykazał zwiększoną liczbę punktów lęku (41). Mając na uwadze te ustalenia, należy rozważyć parametry psychologiczne, które powinny kierować terapią u kobiet z NCCAH, a w kontekście podejścia zorientowanego na pacjenta należy zaoferować diagnozę psychologiczną i wsparcie.
wnioski
NCCAH jest uważany za częstszą i łagodniejszą formę CAH z powodu zachowania 20-50% aktywności enzymu. Większość pacjentów może zasięgnąć porady lekarza na każdym etapie życia z powodu objawów związanych z nadmiarem androgenów. Należą do nich przedwczesny pubarche, szybki wzrost, hirsutyzm, trądzik, nieregularności miesiączkowe lub niepłodność, a także wiele innych mniej widocznych objawów. Wczesne poranne wartości wyjściowe 17 OHP jako dobry wstępny test przesiewowy i dalsza ocena z stymulacją ACTH, a w przypadku wyników granicznych, testy genetyczne są zalecane. Zasadniczo podczas obserwacji należy stosować androstendion, a nie poziomy 17 OHP.
decyzja o leczeniu powinna być oparta na ocenie faktów i powinna być zgodna z indywidualnym podejściem. Leczenie nie zawsze jest wskazane, chyba że pacjent jest objawowy, na przykład dzieci z wczesnym początkiem i szybkim postępem włosów łonowych i ciała, szybkim wzrostem i/lub zaawansowaniem szkieletowym lub kobiety z oligomenorrhea, trądzikiem, hirsutyzmem, niepłodnością lub kombinacją tych i innych wyżej wymienionych objawów. Poradnictwo genetyczne jest zdecydowanie zalecane u kobiet NCCAH, które chcą zajść w ciążę, a także genotypowanie ojca. Ogólne postępowanie pacjenta obejmuje dodatkowo Postępowanie w przypadku prawdopodobnych powikłań leczenia glukokortykoidami lub związanych z metabolizmem objawów choroby. Wreszcie, biorąc pod uwagę, że wiele kwestii terapeutycznych związanych z odpowiednim leczeniem tych pacjentów nie zostało jeszcze wyjaśnionych, bardzo ważne jest, aby lekarz prowadzący był na bieżąco ze wszystkimi zmianami w tej dziedzinie i uwzględniał nowe dane w swojej praktyce klinicznej. Z pewnością konieczne są dalsze badania kliniczne w tej dziedzinie.
podsumowując, dla kobiety z NCCAH idealne podejście jest szyte na miarę, obejmujące płynne przejście jej zarządzania, gdy zostanie skierowana z pediatry do endokrynologa dorosłych, wraz z leczeniem zorientowanym na objawy, które będzie towarzyszyć jej przez całe życie. Biorąc pod uwagę wiele czynników wpływających na układ hiperandrogenny, wskazane jest, aby zachęcić pacjentów do przyjęcia zdrowszego stylu życia poprzez poprawę swoich nawyków żywieniowych, zwiększenie ćwiczeń fizycznych i dążenie do redukcji masy ciała. Ponadto w niektórych przypadkach wsparcie psychologiczne jest często korzystne. Ponadto specjaliści w dziedzinach związanych z leczeniem tego zaburzenia, tacy jak dermatolodzy, ginekolodzy i psycholodzy, potrzebują dogłębnej wiedzy na temat postępowania z podejrzanymi lub już zdiagnozowanymi przypadkami NCCAH. Podsumowując, pamiętajmy o wnikliwym cytacie amerykańskiej astronom very Cooper Rubin: „nauka rozwija się najlepiej, gdy obserwacje zmuszają nas do zmiany naszych uprzedzeń.”
wkład autora
wszyscy wymienieni autorzy wnieśli znaczący, bezpośredni i intelektualny wkład w pracę i zatwierdzili ją do publikacji.
Oświadczenie o konflikcie interesów
autorzy oświadczają, że badanie zostało przeprowadzone przy braku jakichkolwiek relacji handlowych lub finansowych, które mogłyby być interpretowane jako potencjalny konflikt interesów.
1. Speiser PW, biały PC. Wrodzony przerost nadnerczy. N Engl J Med. (2003) 349:776–88. doi: 10.1056/NEJMra021561
PubMed Abstract | CrossRef Full Text/Google Scholar
2. Turcu AF, Auchus RJ. Adrenal steroidogenesis and congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. (2015) 44:275–96. doi: 10.1016/j.ecl.2015.02.002
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Speiser PW. Nonclassic adrenal hyperplasia. Rev Endocr Metab Disord. (2009) 10:77–82. doi: 10.1007/s11154-008-9097-x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Gidlöf S, Falhammar H, Thilén A, von Döbeln U, Ritzén m, Wedell A, et al. Sto lat wrodzonego przerostu nadnerczy w Szwecji: retrospektywne, oparte na populacji badanie kohortowe. Cukrzyca Lancet Endocrinol. (2013) 1:35–42. doi: 10.1016/S2213-8587(13)70007-X
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, New MI. Wysoka częstotliwość nieklasycznego niedoboru 21-hydroksylazy steroidowej. Am J Hum Genet. (1985) 37:650–67.
PubMed Abstract | Google Scholar
6. Wilson RC, Nimkarn S, Dumic M, Obeid J, Azar MR, Azar M, et al. Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Mol Genet Metab. (2007) 90:414–21. doi: 10.1016/j.ymgme.2006.12.005
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Miller WL. Clinical review 54: genetics, diagnosis, and management of 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1994) 78:241–6. doi: 10.1210/jcem.78.2.8106606
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Witchel SF, Azziz R. Nonclassic congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2010) 2010:625105. doi: 10.1186/1687-9856-2010-625105
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Wilson RC, Mercado AB, Cheng KC, New MI. Niedobór 21-hydroksylazy steroidowej: genotyp może nie przewidywać fenotypu. J Clin Endocrinol Metab. (1995) 80:2322–9. doi: 10.1210 / jc.80.8.2322
CrossRef Full Text | Google Scholar
10. Biały PC. Badania przesiewowe noworodków pod kątem wrodzonego przerostu nadnerczy. Nat Rev Endocrinol. (2009) 5:490–8. doi: 10.1038 / nrendo.2009.148
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Kohn B, Levine LS, Pollack MS, Pang S, Lorenzen F, Levy D, et al. Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. (1982) 55:817–27. doi: 10.1210/jcem-55-5-817
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Moran C, Azziz R, Carmina E, Dewailly D, Fruzzetti F, Ibañez L i in. nieklasyczny rozrost nadnerczy z niedoborem Hydroxylazy 21 jest zaburzeniem postępującym: badanie wieloośrodkowe. Am J Obstet Gynecol. (2000) 183:1468-74. doi: 10.1067/mob.2000.108020
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Azziz R, La, Knochenhauer ES, Dewailly D, Fox L, Boots LR. Screening for 21-hydroxylase-deficient nonclassic nadnerczy hyperplasia among hyperandrogenic women: a prospective study. Fertil Steril. (1999) 72:915–25. doi: 10.1016/s0015-0282(99)00383-0
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Livadas S, Dracopoulou M, Dastamani a, Sertedaki a, Maniati-Christidi M, Magiakou A-M, et al. Spektrum badań klinicznych, hormonalnych i molekularnych u 280 osób z nieklasycznym wrodzonym rozrostem nadnerczy spowodowanym mutacjami genu CYP21A2. Clin Endocrinol (Oxf). (2015) 82:543–9. doi: 10.1111 / cen.12543
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Bidet M, Bellanné-Chantelot C, Galand-Portier m-B, Tardy V, Billaud L, Laborde K, et al. Charakterystyka kliniczna i molekularna kohorty 161 niepowiązanych kobiet z nieklasycznym wrodzonym rozrostem nadnerczy spowodowanym niedoborem 21-hydroksylazy I 330 członków rodziny. J Clin Endocrinol Metab. (2009) 94:1570–8. doi: 10.1210 / jc.2008-1582
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Escobar-Morreale HF, Sanchón R, San Millán JL. Prospektywne badanie częstości występowania nieklasycznego wrodzonego przerostu nadnerczy u kobiet z objawami i objawami hiperandrogennymi. J Clin Endocrinol Metab. (2008) 93:527–33. doi: 10.1210 / jc.2007-2053
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Dewailly D, Vantighem-Haudiquet MC, Sainsard C, Buvat J, Cappoen JP, Ardaens K, et al. Fenotypy kliniczne i biologiczne w późnym niedoborze 21-hydroksylazy. J Clin Endocrinol Metab. (1986) 63:418–23. doi: 10.1210 / jcem-63-2-418
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Gönç EN, Ozön ZA, Alikaşifoglu A, Engiz O, Bulum B, Kandemir N. (2011). Czy podstawowa surowica progesteronu 17-OH jest wiarygodnym parametrem do przewidywania nieklasycznego wrodzonego przerostu nadnerczy w przedwczesnym nadnerczu? Turk J Pediatr. 53:274-80.
PubMed Abstract | Google Scholar
19. Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, et al. Wrodzony przerost nadnerczy due to steroid 21-hydroxylase deficiency: an endocrine Society clinical practice guideline. J Clin Endokrynol Metab. (2010) 95: 4133-60. doi: 10.1210 / jc.2009-2631
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Dacou-Voutetakis C, Dracopoulou M. High incidence of molecular defects of the CYP21 gene in patients with premature adrenarche. J Clin Endocrinol Metab. (1999) 84:1570–4. doi: 10.1210/jc.84.5.1570
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Ambroziak U, Kepczynska-Nyk A, Kuryłowicz A, Małunowicz EM, Wójcicka A, Miśkiewicz P, et al. The diagnosis of nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency, based on serum basal or post-ACTH stimulation 17-hydroxyprogesterone, can lead to false-positive diagnosis. Clin Endocrinol (Oxf). (2016) 84:23–9. doi: 10.1111/cen.12935
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Ambroziak U, Kepczynska-Nyk A, Kuryłowicz A, Wysłouch-Cieszynska A, Małunowicz EM, Bartoszewicz Z, et al. LC-MS/MS improves screening towards 21-hydroxylase deficiency. Gynecol Endocrinol. (2015) 31:296–300. doi: 10.3109/09513590.2014.994599
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Keefe CC, Goldman MM, Zhang K, Clarke N, Reitz RE, Welt CK. Jednoczesny pomiar trzynastu hormonów steroidowych u kobiet z zespołem policystycznych jajników i kobiet kontrolnych przy użyciu chromatografii cieczowej-tandemowej spektrometrii masowej. PLoS 1. (2014) 9:e93805. doi: 10.1371 / dziennik.pone.0093805
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Ray ja, Kushnir MM, Yost RA, Rockwood AL, Wayne Meikle A. performance enhancement in the measurement of 5 endogenne steroids by LC-MS / MS combined with differential Ion mobility spectrometry. Clin Chim Acta. (2015) 438:330-6. doi: 10.1016/j.cca.2014 roku.07.036
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Stoupa a, Gonzalez Brisegno L, Pinto G, Samara-Boustani D, Thalassinos C, Flechtner I, et al. Inadequate cortisol response to the tetracosactide (Synacthen®) test in non-classic wrodzony przerost nadnerczy: an exception to the rule? Horm Res Paediatr. (2015) 83: 262-7. doi: 10.1159/000369901
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. (1992) 90:584–95. doi: 10.1172/JCI115897
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Temeck JW, Pang Sy, Nelson C, New MI. Wady genetyczne steroidogenezy w przedwczesnym pubarche. J Clin Endocrinol Metab. (1987) 64:609–17. doi: 10.1210 / jcem-64-3-609
PubMed Abstract | CrossRef Full Text | Google Scholar
28. Voutilainen R, Jääskeläinen J. Premature adrenarche: etiology, clinical findings, and consequences. J Steroid Biochem Mol Biol. (2015) 145:226–36. doi: 10.1016 / j.jsbmb.2014.06.004
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Oberfield SE, Sopher AB, Gerken AT. Approach to the girl with early onset of pubic hair. J Clin Endocrinol Metab. (2011) 96:1610–22. doi: 10.1210/jc.2011-0225
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Janus D, Wojcik M, Malunowicz E, Starzyk JB. Przypadek nawracających zrostów wargowych u 15-miesięcznego dziecka z bezobjawowym wrodzonym rozrostem nadnerczy spowodowanym niedoborem 21-hydroksylazy. J Pediatr Endocrinol Metab. (2012) 25:1017–21. doi: 10.1515/jpem-2012-0190
PubMed Abstract | CrossRef Full Text/Google Scholar
31. Gopal-Kothandapani JS, Petkar A, O ’ Shea E, Banerjee I. włosy okołoodbytnicze jako niezwykła prezentacja nieklasycznego wrodzonego przerostu nadnerczy. (2013) 2013:bcr2013010123. doi: 10.1136/bcr-2013-010123
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Falhammar H, Nordenström A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine. (2015) 50:32–50. doi: 10.1007/s12020-015-0656-0
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Chrousos GP, Loriaux DL, Mann DL, Cutler GB. Late-onset 21-hydroxylase deficiency mimicking idiopathic hirsutism or polycystic ovarian disease. Ann Intern Med. (1982) 96:143–8. doi: 10.7326/0003-4819-96-2-143
CrossRef Full Text | Google Scholar
34. Witchel SF. Non-classic congenital adrenal hyperplasia. Steroids. (2013) 78:747–50. doi: 10.1016/j.steroids.2013.04.010
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Carmina E, Rosato F, Jannì A, Rizzo M, Longo RA. Extensive clinical experience: relative prevalence of different androgen excess disorders in 950 women referred because of clinical hyperandrogenism. J Clin Endocrinol Metab. (2006) 91:2–6. doi: 10.1210/jc.2005-1457
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Moran C, Azziz R, Weintrob N, Witchel SF, Rohmer V, Dewailly D, et al. Wyniki reprodukcyjne u kobiet z nieklasycznym rozrostem nadnerczy z niedoborem 21-hydroksylazy. J Clin Endocrinol Metab. (2006) 91:3451–6. doi: 10.1210 / jc.2006-0062
PubMed Abstract | CrossRef Full Text | Google Scholar
37. Levin JH, Carmina E, Lobo RA. Czy niewłaściwe wydzielanie gonadotropin u pacjentów z zespołem policystycznych jajników jest podobne do u pacjentów z wrodzonym rozrostem nadnerczy u dorosłych? Fertil Steril. (1991) 56:635–40. doi: 10.1016/S0015-0282(16)54592-0
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Carmina E, Lobo RA. Ovarian suppression reduces clinical and endocrine expression of late-onset congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Fertil Steril. (1994) 62:738–43. doi: 10.1016/S0015-0282(16)56998-2
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Ludwig E. Klasyfikacja rodzajów łysienia androgenowego (łysienie powszechne) występujących u płci żeńskiej. Br J Dermatol. (1977) 97:247–54. doi: 10.1111 / j. 1365-2133. 1977.tb15179x
PubMed Abstract | CrossRef Full Text | Google Scholar
40. Trakakis e, Papadavid e, Dalamaga m, Koumaki D,Stavrianeas N, Rigopoulos D, et al. Częstość występowania nieklasycznego wrodzonego przerostu nadnerczy z powodu niedoboru 21-hydroksylazy u greckich kobiet z trądzikiem: badanie przekrojowe prowadzone w szpitalu. J Eur Acad Dermatol Venereol. (2013) 27:1448–51. doi: 10.1111 / j.1468-3083.2012.04613.x
PubMed Abstract | CrossRef Full Text | Google Scholar
41. Arlt W, Willis DS, Wild SH, Krone N, Doherty EJ, Hahner S, et al. Stan zdrowia dorosłych z wrodzonym rozrostem nadnerczy: badanie kohortowe z udziałem 203 pacjentów. J Clin Endocrinol Metab. (2010) 95:5110–21. doi: 10.1210 / jc.2010-0917
PubMed Abstract | CrossRef Full Text | Google Scholar
42. Saygili F, Oge a, Yilmaz C. hiperinsulinemia i niewrażliwość na insulinę u kobiet z nieklasycznym wrodzonym rozrostem nadnerczy z powodu niedoboru 21-hydroksylazy: związek między poziomem leptyny w surowicy a przewlekłą hiperinsulinemią. Horm Res. (2005) 63: 270-4. doi: 10.1159/000086363
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Speiser PW, Serrat J, New MI, Gertner JM. Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1992) 75:1421–4. doi: 10.1210/jc.75.6.1421
PubMed Abstract | CrossRef Full Text | Google Scholar
44. Han TS, Stimson RH, Rees DA, Krone N, Willis DS, Conway GS i in. Schemat leczenia glikokortykosteroidami i wyniki zdrowotne u dorosłych z wrodzonym rozrostem nadnerczy. Clin Endocrinol. (2013) 78:197–203. doi: 10.1111 / cen.12045
PubMed Abstract | CrossRef Full Text | Google Scholar
45. Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. stosowanie doustnych kortykosteroidów a ryzyko złamań. J Bone Miner Res. (2000) 15: 993-1000. doi: 10.1359 / jbmr.2000.15.6.993
PubMed Abstract | CrossRef Full Text | Google Scholar
46. Nebesio TD, Renbarger JL, Nabhan ZM, Ross SE, Slaven JE, Li L, et al. Differential effects of hydrocortisone, prednisone, and dexamethasone on hormonal and pharmacokinetic profiles: a pilot study in children with congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2016) 2016:17. doi: 10.1186 / s13633-016-0035-5
PubMed Abstract | CrossRef Full Text | Google Scholar
47. Weintrob N, Dickerman Z, Sprecher e, Galatzer a, Pertzelan A. nieklasyczny niedobór 21-hydroksylazy w okresie niemowlęcym i dziecięcym: wpływ czasu rozpoczęcia terapii na okres dojrzewania i ostateczny wzrost. Eur J Endocrinol. (1997) 136:188–95. doi: 10.1530 / eje.0.1360188
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Bonfig W, Bechtold S, Schmidt H, Knorr D, Schwarz HP. Reduced final height outcome in congenital adrenal hyperplasia under prednisone treatment: deceleration of growth velocity during puberty. J Clin Endocrinol Metab. (2007) 92:1635–9. doi: 10.1210/jc.2006-2109
PubMed Abstract | CrossRef Full Text | Google Scholar
49. Dörr HG. Wzrost u pacjentów z klasycznym wrodzonym rozrostem nadnerczy spowodowanym niedoborem 21-hydroksylazy. Horm Res. (2007) 68 (Suppl 5): 93-9. doi: 10.1159/000110587
PubMed Abstract | CrossRef Full Text/Google Scholar
50. Hoepffner w, Kaufhold a, Willgerodt H, Keller E. pacjenci z klasycznym wrodzonym przerostem nadnerczy spowodowanym niedoborem 21-hydroksylazy mogą osiągnąć swój docelowy wzrost: doświadczenie Lipska. Horm Res. (2008) 70: 42-50. doi: 10.1159/000129677
PubMed Abstract | CrossRef Full Text | Google Scholar
51. van der Kaay D, van den Akker E. Ultralow-dose dexamethasone to preserve endogenous cortisol stress response in nonclassical congenital adrenal hyperplasia: a new promising treatment. Int J Endocrinol Metab. (2014) 12:e14657. doi: 10.5812/ijem.14657
CrossRef Full Text | Google Scholar
52. Lin-Su K, Vogiatzi MG, Marshall I, Harbison MD, Macapagal MC, Betensky B, et al. Treatment with growth hormone and luteinizing hormone releasing hormone analog improves final adult height in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. (2005) 90:3318–25. doi: 10.1210/jc.2004-2128
PubMed Abstract | CrossRef Full Text | Google Scholar
53. McCann-Crosby B, Chen M-J, Lyons SK, Lin Y, Axelrad M, Dietrich JE, et al. Nonclassical congenital adrenal hyperplasia: cele leczenia i przejścia. Pediatr Endocrinol Rev. (2014)12: 224-38.
PubMed Abstrakt/Google Scholar
54. Ogilvie CM, Crouch NS, Rumsby G, Creighton SM, Liao L-M, Conway GS. Wrodzony przerost nadnerczy u dorosłych: przegląd zagadnień medycznych, chirurgicznych i psychologicznych. Clin Endocrinol. (2006) 64:2–11. doi: 10.1111 / j.1365-2265.2005.02410.x
PubMed Abstract | CrossRef Full Text | Google Scholar
55. Mah PM, Jenkins RC, Rostami-Hodjegan A, Newell-Price J, Doane A, Ibbotson V, et al. Weight-related dosing, timing and monitoring hydrocortisone replacement therapy in patients with adrenal insufficiency. Clin Endocrinol. (2004) 61:367–75. doi: 10.1111/j.1365-2265.2004.02106.x
PubMed Abstract | CrossRef Full Text | Google Scholar
56. Diaz A, Laufer Mr Breech LLAA z PC na a, Care AC na O i GC na AH. Miesiączka u dziewcząt i młodzieży: wykorzystanie cyklu miesiączkowego jako znaku życiowego. Pediatria. (2006) 118:2245–50. doi: 10.1542 / peds.2006-2481
CrossRef Full Text | Google Scholar
57. Merke DP, Poppas DP. Postępowanie u młodzieży z wrodzonym rozrostem nadnerczy. cukrzyca lancet Endocrinol. (2013) 1:341–52. doi: 10.1016/S2213-8587(13)70138-4
PubMed Abstract | CrossRef Full Text | Google Scholar
58. Verma S, Green-Golan L, VanRyzin C, Drinkard B, Mehta SP, Weise M, et al. Adrenomedullary function in patients with nonclassic congenital adrenal hyperplasia. Horm Metab Res. (2010) 42:607–12. doi: 10.1055/s-0030-1253385
PubMed Abstract | CrossRef Full Text | Google Scholar
59. New MI. Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2006) 91:4205–14. doi: 10.1210/jc.2006-1645
PubMed Abstract | CrossRef Full Text | Google Scholar
60. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2018) 103:4043–88. doi: 10.1210/jc.2018-01865
PubMed Abstract | CrossRef Full Text | Google Scholar
61. Kruse B, Riepe FG, Krone N, Bosinski HAG, Kloehn S, Partsch CJ, et al. Congenital adrenal hyperplasia – how to improve the transition from adolescence to adult life. Exp Clin Endocrinol Diabetes. (2004) 112:343–55. doi: 10.1055/s-2004-821013
PubMed Abstract | CrossRef Full Text | Google Scholar
62. Gastaud F, Bouvattier C, Duranteau L, Brauner R, Thibaud E, Kutten F, et al. Zaburzenia czynności seksualnych i rozrodczych u kobiet z klasycznymi postaciami wrodzonego przerostu nadnerczy. J Clin Endocrinol Metab. (2007) 92:1391–6. doi: 10.1210 / jc.2006-1757
PubMed Abstract | CrossRef Full Text | Google Scholar
63. Mercè Fernández-Balsells M, Muthusamy K, Smushkin G,Lampropulos JF, Elamin MB, Abu Elnour NO, et al. Prenatalne zastosowanie deksametazonu w zapobieganiu wirylizacji u kobiet w ciąży zagrożonych klasycznym wrodzonym rozrostem nadnerczy z powodu niedoboru 21-hydroksylazy (CYP21A2): przegląd systematyczny i metaanalizy. Clin Endocrinol. (2010) 73:436–44. doi: 10.1111 / j. 1365-2265. 2010. 03826.x
CrossRef Full Text | Google Scholar
64. Bidet M, Bellanné-Chantelot C, Galand-Portier MB, Golmard JL, Tardy V, Morel Y, et al. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2010) 95:1182–90. doi: 10.1210/jc.2009-1383
PubMed Abstract | CrossRef Full Text | Google Scholar
65. Birnbaum MD, Rose LI. Late onset adrenocortical hydroxylase deficiencies associated with menstrual dysfunction. Obstet Gynecol. (1984) 63:445–51.
PubMed Abstract | Google Scholar
66. Cuhaci N, Aydin C, Yesilyurt A, Pinarli FA, Ersoy R, Cakir B. Nonclassical congenital adrenal hyperplasia and pregnancy. Case Rep Endocrinol. (2015) 2015:296924. doi: 10.1155/2015/296924
PubMed Abstract | CrossRef Full Text | Google Scholar
67. Deneux C, Tardy V, Anne D, Mornet E, Billaud L, Charron D, et al. Korelacja fenotypowo-genotypowa u 56 kobiet z nieklasycznym wrodzonym rozrostem nadnerczy spowodowanym niedoborem 21-hydroksylazy. J Clin Endocrinol Metab. (2001) 86:207–13. doi: 10.1210 / jcem.86.1.7131
PubMed Abstract | CrossRef Full Text | Google Scholar
68. Simonetti L, Bruque CD, Fernández CS, Benavides-Mori B, Delea m, Kolomenski JE, et al. Aktualizacja mutacji CYP21A2: kompleksowa analiza baz danych i opublikowanych wariantów genetycznych. Hum Mutat. (2018) 39:5–22. doi: 10.1002 / humu.23351
PubMed Abstract | CrossRef Full Text | Google Scholar
69. OMIM Entry – * 613815 – CYTOCHROME P450 FAMILY 21 SUBFAMILY A POLYPEPTIDE 2; CYP21A2.
Google Scholar
70. Miller WL, Witchel SF. Prenatal treatment of congenital adrenal hyperplasia: risks outweigh benefits. Am J Obstet Gynecol. (2013) 208:354–9. doi: 10.1016/j.ajog.2012.10.885
PubMed Abstract | CrossRef Full Text | Google Scholar
71. Lekarev O, New MI. Adrenal disease in pregnancy. Best Pract Res Clin Endocrinol Metab. (2011) 25:959–73. doi: 10.1016/j.beem.2011.08.004
PubMed Abstract | CrossRef Full Text | Google Scholar
72. New MI, Abraham M, Yuen T, Lekarev O. An update on prenatal diagnosis and treatment of congenital adrenal hyperplasia. Semin Reprod Med. (2012) 30:396–9. doi: 10.1055/s-0032-1324723
PubMed Abstract | CrossRef Full Text | Google Scholar
73. Kazmi D, Bailey J, Yau M, Abu-Amer W, Kumar A, Low M, et al. New developments in prenatal diagnosis of congenital adrenal hyperplasia. J Steroid Biochem Mol Biol. (2017) 165:121–3. doi: 10.1016/j.jsbmb.2016.06.016
PubMed Abstract | CrossRef Full Text | Google Scholar
74. Clayton PE, Miller WL, Oberfield SE, Ritzén EM, Sippell WG, Speiser PW, et al. Oświadczenie konsensusowe w sprawie niedoboru 21-hydroksylazy od Europejskiego Towarzystwa Endokrynologii pediatrycznej i lawson wilkins pediatric endocrine society. Horm Res. (2002) 58: 188-95. doi: 10.1159/000065490
PubMed Abstract | CrossRef Full Text/Google Scholar
75. Nandagopal R, Sinaii N, Avila NA, Van Ryzin C, Chen w, Finkielstain GP, et al. Profilowanie fenotypowe rodziców z tajemniczym nieklasycznym wrodzonym rozrostem nadnerczy: odkrycia w 145 niepowiązanych rodzinach. Eur J Endocrinol. (2011) 164:977–84. doi: 10.1530/EJE-11-0019
PubMed Abstract | CrossRef Full Text/Google Scholar
76. Takasu N, Nakachi K, Higa H. rozwój nadczynności tarczycy Gravesa-Basedowa spowodował przełom nadnerczy u pacjenta z wcześniej nierozpoznanym nieklasycznym niedoborem 21-hydroksylazy. Intern Med. (2010) 49:1395–400. doi: 10.2169 / internalmedicine.49.3573
PubMed Abstract | CrossRef Full Text | Google Scholar
77. Meyer-Bahlburg HFL, Dolezal C, Baker SW, Carlson AD, Obeid JS, New MI. Prenatal androgenization affects gender-related behavior but not gender identity in 5-12-year-old girls with congenital adrenal hyperplasia. Arch Sex Behav. (2004) 33:97–104. doi: 10.1023/B:ASEB.0000014324.25718.51
CrossRef Full Text | Google Scholar
78. Meyer-Bahlburg HFL, Dolezal C, Baker SW, New MI. Orientacja seksualna u kobiet z klasycznym lub nieklasycznym wrodzonym przerostem nadnerczy jako funkcja prenatalnego nadmiaru androgenów. Arch Sex Behavior. (2008) 37:85–99. doi: 10.1007 / s10508-007-9265-1
PubMed Abstract / CrossRef Full Text / Google Scholar