pierwotna struktura białka jest zdefiniowana jako sekwencja aminokwasów, z których się składa. Sekwencja ta ostatecznie określa kształt, jaki przyjmuje białko, zgodnie z ograniczeniami przestrzennymi dotyczącymi ułożenia atomów w białku, właściwościami chemicznymi składowych reszt aminokwasowych i środowiskiem białka.
wiązania peptydowe, które łączą reszt aminokwasowych w polipeptydzie, powstają w reakcji kondensacji między kwaśną grupą karboksylową jednego aminokwasu a zasadową grupą aminową innego aminokwasu. W kontekście peptydu grupę amidową (CO–NH) określa się mianem grupy peptydowej.
kluczowe dla zrozumienia struktury białka jest znajomość struktury wiązania peptydowego. Linus Pauling, w latach trzydziestych XX wieku, użył dyfrakcji rentgenowskiej do zbadania natury wiązania peptydowego utworzonego między dwoma aminokwasami. Poinformował, że grupa peptydowa (CO–NH) ma sztywną strukturę płaską. Struktura ta jest wynikiem oddziaływań między elektronami wiązania podwójnego grupy karbonylowej a elektronami wiązania C-N (fig. 2), tak że elektrony te nabywają częściowe (około 40%) właściwości wiązania podwójnego.

efekt ten jest przykładem rezonansu, który można traktować jako dzielenie elektronów między wiązaniami. Ponieważ pojedyncze wiązania między dwoma atomami są dłuższe niż podwójne wiązania między tymi samymi dwoma atomami, długości wiązań C–N I C=O w grupie peptydowej różnią się od tych obserwowanych dla tych wiązań w innych kontekstach, w których rezonans nie występuje. Tak więc cząstkowy charakter wiązania podwójnego C – N w grupie peptydowej oznacza, że Wiązanie to jest krótsze niż przewidywałoby się dla pojedynczego wiązania C–N, podczas gdy Wiązanie C=O, mające cząstkowy charakter pojedynczego wiązania ze względu na rezonans, jest dłuższe niż przewidywalne dla podwójnego wiązania C=O. Długości wiązania w grupie peptydowej przedstawiono na fig. 3. Porównaj Wiązanie C-N grupy peptydowej z wiązaniem między N i Ca (atom C, do którego przyłączona jest grupa aminowa i grupa karboksylowa).

możliwe są dwie konformacje planarnego wiązania peptydowego: w grupie peptydów trans Atomy Ca znajdują się po przeciwnych stronach wiązania peptydowego (Fig.3a), a w grupie peptydów cis Atomy Ca znajdują się po tej samej stronie wiązania peptydowego (Fig. 3b).
-
biorąc pod uwagę układ przestrzenny i bliskość atomów w konformacjach cis i trans wiązania peptydowego, która konformacja według Ciebie byłaby preferowana?
-
konformacja trans byłaby energetycznie korzystniejsza niż konformacja cis, ponieważ minimalizuje przeszkody steryczne.
ogólnie rzecz biorąc, wiązania peptydowe są w konformacji trans. Jednak formy cis mogą występować w wiązaniach peptydowych, które poprzedzają resztę prolinową. W takich przypadkach forma cis jest bardziej stabilna niż zwykle, ponieważ łańcuch boczny proline oferuje mniej przeszkód. Niemniej jednak, wiązania peptydowe cis występują tylko w około 10% przypadków wiązań peptydowych poprzedzających pozostałości proliny.
mając na uwadze planarny charakter grupy peptydowej, można zauważyć, że łańcuch polipeptydowy ma szkielet, który składa się z szeregu sztywnych planarnych grup peptydowych połączonych atomami Ca. 4 przedstawia część polipeptydu z dwiema planarnymi grupami peptydowymi w konformacji trans. Należy zauważyć, że chociaż rotacja nie jest dozwolona w odniesieniu do wiązań peptydowych, istnieje możliwość rotacji wokół wiązań Ca–N i Ca–C. Kąty obrotu, zwane kątami skrętu, dotyczące tych wiązań określają konformację szkieletu polipeptydu. Kąty skrętu wokół wiązań Ca–N i Ca–C określane są jako ɸ (phi) i ψ. (psi), odpowiednio i są one zdefiniowane jako 180°, gdy polipeptyd jest w rozszerzonej konformacji płaskiej, jak pokazano na fig.
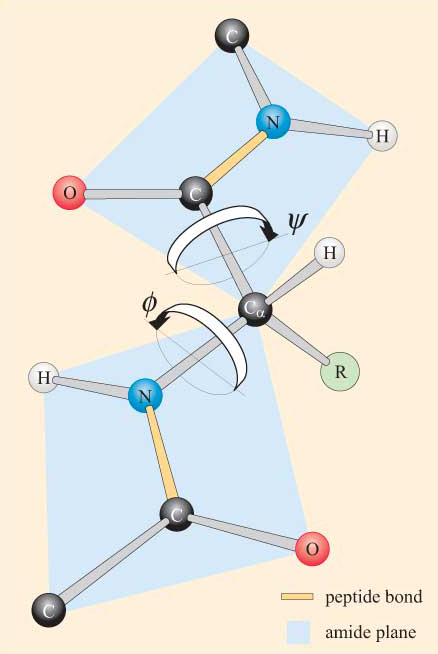
nie zdziwisz się, że ograniczenia steryczne dotyczą ɸ i ψ.
w wyniku tych ograniczeń sterycznych dozwolone są tylko niektóre wartości ɸ i ψ, a więc konformacje peptydu, podczas gdy inne nie.
możliwe jest obliczenie tych dopuszczalnych wartości dla danej pozostałości w kontekście polipeptydu. Obliczenia te wykonuje się, określając najpierw odległości między wszystkimi niewsprzęgającymi atomami w dwóch sąsiednich grupach peptydowych (takich jak te na fig.4) przy wszystkich możliwych wartościach ψ i ψ. Najłatwiej jest to zrobić dla polipeptydu zawierającego tylko jeden rodzaj aminokwasu. Wykres konformacyjny ɸ względem ψ dla konkretnej pozostałości jest znany jako wykres Ramachandrana (od jego wynalazcy, G. N. Ramachandrana). Taki wykres pozwala nam zidentyfikować te konformacje (tj. dla określonej wartości ψ i ψ), które są sterycznie korzystne lub niekorzystne (jak na fig.5), według następujących kryteriów:
-
tam, gdzie nie ma konfliktu między promieniami van der Waalsa nie wiążących się atomów, konformacja jest „dozwolona”. Konformacje te leżą na niebieskich obszarach na fig.
-
konformacje wymagające międzyatomowych odległości na granicy tego, co jest dopuszczalne są definiowane jako konformacje „granicy zewnętrznej”. Leżą na terenach zielonych na rysunku 5.
-
konformacje teoretyczne, które wymagają, aby dowolne dwa niezwiązane Atomy były bliżej siebie niż pozwalają na to promienie van der Waalsa, są sterycznie „zabronione”. Leżą one w białych obszarach na rysunku 5.
zauważ, że wartości ψ i ψ Na Rysunku 5 wahają się od −180º do +180º. Obrócenie grupy peptydowej przez 360º oczywiście przywróci ją do pozycji wyjściowej, a −180º i +180º odpowiadają tej samej pozycji. Tak więc zielony pasek w lewym dolnym rogu wykresu na fig. 5 sąsiaduje z polem w lewym górnym rogu.

-
użyj rysunku 5, aby określić, czy następujące wartości ɸ i ψ są sterycznie korzystne lub niekorzystne: (a) ɸ = 90º i ψ = 90º; (b) ɸ = −90º i ψ = 90º.
-
(a) ; B) korzystne.
Ramachandran może być skonstruowany dla polimerów każdego z 20 aminokwasów. Istotne jest, aby zauważyć, że wykresy Ramachandranu dla wielu reszt aminokwasowych są na ogół bardzo podobne, mając tylko trzy regiony o korzystnych lub tolerowanych konformacjach (oznaczone 1-3 na wykresie dla Poli-l-alaniny na fig.5). Różnice jednak występują. Na przykład, gdy łańcuch boczny (R na fig. 4) jest rozgałęziony w pobliżu Ca, jak w przypadku treoniny, zajmuje on więcej miejsca w pobliżu szkieletu peptydowego i ogranicza zbliżanie się atomów w sąsiednich grupach peptydowych. W rezultacie dozwolone konformacje (kąty ψ i ψ) są bardziej ograniczone dla polipeptydów aminokwasów rozgałęzionych.
-
prolina różni się również od innych aminokwasów pod względem dozwolonych konformacji, a dla poliproliny tolerowane są tylko ɸ wartości od −85º do −35º. Myśląc o strukturze proliny, jak wytłumaczyć ten stosunkowo wąski zakres dopuszczalnych wartości?
-
łańcuch boczny proliny jest kowalencyjnie związany z N grupy aminowej, więc w poliprolinie będzie mniejsza swoboda rotacji wokół wiązania Ca-n niż w przypadku innych aminokwasów. W związku z tym dopuszczalne wartości ɸ będą stosunkowo ograniczone w porównaniu z innymi aminokwasami.
-
Rysunek 6 przedstawia wykres Ramachandranu dla reszt glicyny w łańcuchu polipeptydowym. Regiony są oznaczone kolorami, jak na rysunku 5. Co możesz powiedzieć o konformacjach, które przyjmuje glicyna? Rozważ strukturę glicyny. Dlaczego glicyna różni się od innych pozostałości pod względem konformacji?
-
glicyna ma znacznie większą swobodę konformacyjną niż inne reszty aminokwasowe, ponieważ jest mniej sterycznie utrudniona.

wykresy Ramachandranu na fig. 5 i 6 zostały wygenerowane odpowiednio dla l-alaniny i L-glicyny na podstawie dozwolonych i zewnętrznych odległości granicznych dla kontaktów międzyatomowych, określonych na podstawie znanych wartości promieni van der Waalsa atomów (Tabela 1).
Tabela 1 odległości Van der Waalsa dla kontaktów międzyatomowych.
| Typ kontaktu | normalnie dozwolone/Å | granica zewnętrzna/Å | |
|---|---|---|---|
| H···H | 2.0 | 1.9 |
3.0 |
są więc raczej predykcyjne niż rzeczywiste wykresy konformacyjne. Możemy oczywiście użyć dyfrakcji rentgenowskiej do eksperymentalnego określenia „rzeczywistych” wartości ɸ i ψ Dla reszt w polipeptydzie. Na fig. 7 wartości ψ i ψ dla wszystkich reszt (z wyjątkiem glicyny i proliny) w wielu różnych strukturach zostały określone za pomocą dyfrakcji rentgenowskiej o wysokiej rozdzielczości i wykreślone na wykresie Ramachandrana. Widzimy, że istnieje uderzająca zgodność między przewidywanymi i rzeczywistymi konformacjami. Zauważ jednak, że istnieją pewne pozostałości, których konformacje mapują się z „zakazanymi” obszarami. Większość tych pozostałości mapuje się w regionie między „dozwolonymi” regionami 2 i 3, Około ψ = 0.

-
spójrz ponownie na rysunek 4 i wyobraź sobie, że możesz przekręcić najwyższą grupę peptydową o 180°, tak aby ψ = 0. Jakie grupy są narażone na konflikt w tej konformacji?
-
grupy N–H sąsiadujących grup peptydowych będą ze sobą kolidować, będąc zmuszone do bliskiego Zbliżenia.
konflikt związany z tymi konformacjami może być zażegnany przez niewielki stopień skręcenia wiązania peptydowego. Tak więc, w takich konformacjach Grupa peptydowa jest wykręcona z jej zwykłej konformacji planarnej.
ograniczona liczba „zabronionych” konformacji poszczególnych reszt może być tolerowana w polipeptydzie, jeśli przyjęta konformacja jako całość jest energetycznie korzystna. Polipeptyd ma tendencję do składania się w taki sposób, że przyjmuje najbardziej stabilną konformację. W tej konformacji polipeptyd minimalizuje swoją energię swobodną. W następnych sekcjach przyjrzymy się temu wyższemu poziomowi struktury białka.