den primære strukturen til et protein er definert som sekvensen av aminosyrer som den består av. Denne sekvensen bestemmer til slutt formen som proteinet vedtar, i henhold til de romlige begrensningene på arrangementet av atomene i proteinet, de kjemiske egenskapene til komponentaminosyrerester og proteinets miljø.peptidbindingene som forbinder aminosyrerester i et polypeptid dannes i en kondensasjonsreaksjon mellom den sure karboksylgruppen av en aminosyre og den grunnleggende aminogruppen av en annen aminosyre. I sammenheng med et peptid refereres amidgruppen (CO–NH) til som peptidgruppen.
Avgjørende for en forståelse av proteinstruktur er kunnskap om strukturen av peptidbindingen. Linus Pauling, på 1930-tallet, brukte Røntgendiffraksjon for å undersøke arten av peptidbindingen dannet mellom to aminosyrer. Han rapporterte at peptidgruppen (CO–NH) har en stiv plan struktur. Denne strukturen skyldes interaksjoner mellom elektroner av dobbeltbindingen av karbonylgruppen og De Av C-N-bindingen (Figur 2) slik at sistnevnte oppnår delvise (ca.40%) dobbeltbindingsegenskaper.

denne effekten er et eksempel på resonans som kan betraktes som en deling av elektroner mellom bindinger. Siden enkeltbindinger mellom to atomer er lengre enn dobbeltbindinger mellom de samme to atomer, varierer lengdene Av C–N Og C=O-bindingene i peptidgruppen fra de som observeres for disse bindingene i andre sammenhenger der resonans ikke forekommer. Således betyr den partielle dobbeltbindingskarakteren Av C-N i peptidgruppen at denne bindingen er kortere enn det som ville bli spådd for En c-n enkeltbinding, mens C=O-bindingen, som har en delvis enkeltbindingskarakter på grunn av resonans, er lengre enn det som ville bli spådd for En c=O dobbeltbinding. Bindingslengdene i peptidgruppen er angitt i Figur 3. Sammenlign C – n-bindingen av peptidgruppen med den Mellom N Og Ca (c-atomet som aminogruppen og karboksylgruppen er festet til).

det er to mulige konformasjoner av den plane peptidbindingen: i transpeptidgruppen er Ca-atomer på motsatte sider av peptidbindingen (Figur 3a) og i cis-peptidgruppen er Ca-atomer på samme side av peptidbindingen (Figur 3b).
-
Vurderer romlig arrangement og nærhet av atomene i cis og trans konformasjoner av peptidbindingen, hvilken konformasjon tror du ville bli favorisert?transkonformasjonen ville være energisk gunstigere enn cis-konformasjonen, siden den minimerer sterisk hindring.
generelt sett er peptidbindinger i transkonformasjonen. Cis-former kan imidlertid forekomme i peptidbindinger som går foran en prolinrest. I slike tilfeller er cis-formen mer stabil enn vanlig siden prolin-sidekjeden gir mindre hindring. Ikke desto mindre forekommer cis-peptidbindinger bare i omtrent 10% av forekomster av peptidbindinger før prolinrester.Med tanke på peptidgruppens plane natur, kan en polypeptidkjede ses å ha en ryggrad som består av en serie stive plan peptidgrupper forbundet Med Ca-atomer. Figur 4 viser en del av et polypeptid med to plane peptidgrupper i transkonformasjonen. Merk at selv om rotasjon ikke er tillatt om peptidbindingene, er det potensial for rotasjon rundt Ca-N og Ca–C-bindingene. Rotasjonsvinklene, kalt torsjonsvinkler, om disse bindingene spesifiserer konformasjonen av et polypeptid ryggrad. Vridningsvinklene om Ca–N–og Ca-C-obligasjonene refereres til som ɸ (phi) og ψ (psi), henholdsvis, og de er definert som 180° når polypeptidet er i den utvidede plane konformasjonen, som illustrert i Figur 4.
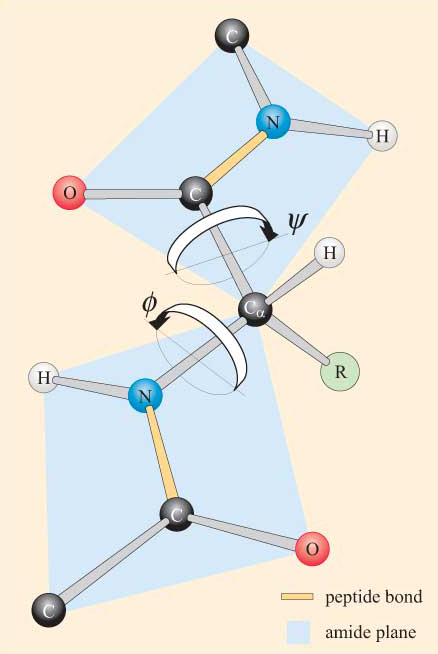
Du vil ikke bli overrasket over å høre at steriske begrensninger gjelder for ɸ og ψ.
som følge av disse steriske begrensningene, er det kun visse verdier av ɸ og ψ, og dermed konformasjoner av peptidet, tillatt mens andre ikke er det.
det er mulig å beregne disse tillatte verdiene for en gitt rest i sammenheng med et polypeptid. Denne beregningen utføres ved først å bestemme avstandene mellom alle ikke-bindende atomer i to nærliggende peptidgrupper (For Eksempel De I Figur 4) ved alle mulige verdier av ɸ og ψ. Det er lettest gjort for et polypeptid som inneholder bare en slags aminosyre. Et konformasjonelt plott av ɸ mot ψ for en bestemt rest er kjent som Et Ramachandran-plott (Etter oppfinneren, G. N. Ramachandran). Et slikt plott gjør det mulig for oss å identifisere de konformasjonene (dvs. for en bestemt verdi av ɸ og ψ) som er sterisk gunstige eller ugunstige (Som I Figur 5), i henhold til følgende kriterier:
-
der det ikke er noen konflikt mellom van Der Waals radier av ikke-bindende atomer, er en konformasjon ’tillatt’. Disse konformasjonene ligger i de blå områdene i Figur 5.
-
Konformasjoner som krever interatomiske avstander ved grensen for det som er tillatt, er definert som ‘ytre grense’ konformasjoner. De ligger i de grønne områdene i Figur 5.Teoretiske konformasjoner som krever at to ikke-bindende atomer er nærmere hverandre enn deres van Der Waals-radier tillater, er strengt forbudt. Disse ligger i de hvite områdene I Figur 5.
Legg merke til at verdiene av ɸ og ψ i Figur 5 varierer fra −180º til +180º. Ved å dreie peptidgruppen til 360º fører den selvfølgelig tilbake til startposisjonen, og −180º og +180º svarer til samme posisjon. Dermed er den grønne stripen nederst i venstre hjørne av tomten i Figur 5 sammenhengende med feltet øverst til venstre.

-
Bruk Figur 5 for å finne ut om følgende verdier av ɸ og ψ er sterically gunstige eller ugunstige: (a) ɸ = 90º og ψ = 90º; (b) ɸ = −90º og ψ = 90º.
-
(a) Ugunstig; (b) gunstig.
Ramachandran plott kan konstrueres for polymerer av hver av de 20 aminosyrene. Det er viktig å merke Seg At Ramachandran-plottene for mange aminosyrerester generelt er svært like, og har bare tre regioner med gunstige eller tolererte konformasjoner(merket 1-3 i plottet for poly-l-alanin I Figur 5). Forskjeller forekommer imidlertid. For eksempel, hvor sidekjeden (R i Figur 4) er forgrenet nær Ca, som i tilfelle av treonin, opptar den mer plass nær peptid-ryggraden og begrenser tilnærmingen til atomer i nabopeptidgruppene. Som følge av dette er tillatte konformasjoner (ɸ og ψ vinkler) mer begrenset for polypeptider av forgrenede aminosyrer.
-
Prolin er også ganske forskjellig fra andre aminosyrer når det gjelder tillatte konformasjoner, og for polyprolin tolereres kun ɸ verdier fra −85º til −35º. Når du tenker på strukturen til proline, hvordan kan du forklare dette relativt smale spekteret av tillatte ɸ?sidekjeden av prolin er kovalent bundet Til n av aminogruppen, så i polyprolin vil det være mindre rotasjonsfrihet om Ca-N-bindingen enn med andre aminosyrer. Følgelig vil tillatte ɸ være relativt begrensede sammenlignet med andre aminosyrer.
-
Figur 6 viser Ramachandran-plottet for glycinrester i en polypeptidkjede. Regionene er fargekodet som I Figur 5. Hva kan du si om konformasjonene som glycin vedtar? Vurder strukturen av glycin. Hvorfor er glycin forskjellig fra de andre rester med hensyn til dens konformasjoner?
-
Glycin har mye større konformasjonsfrihet enn andre aminosyrerester, fordi det er mindre sterisk hindret.

Ramachandran-plottene i Figur 5 og 6 har blitt generert for henholdsvis l-alanin og l-glycin på grunnlag av tillatte og ytre grenseavstander for interatomiske kontakter, bestemt ut fra kjente verdier for van Der Waals radier av atomene (Tabell 1).
Tabell 1 Van Der Waals avstander for interatomiske kontakter.
| Kontakttype | Normalt tillatt / Å | Ytre grense / Å | |
|---|---|---|---|
| H···h | 2.0 | 1.9 |
3.0 |
de er derfor prediktive snarere enn faktiske konformasjonelle plott. Vi kan selvsagt bruke røntgendiffraksjon for å bestemme eksperimentelt de ‘virkelige’ verdiene av ɸ og ψ for rester i et polypeptid. I Figur 7 er det fastsatt høyoppløselig røntgendiffraksjon for alle restrester (med unntak av glysin og prolin) i En Rekke ulike strukturer, og plottet på En Ramachandran-plot. Vi kan se at det er en slående korrespondanse mellom forutsagte og faktiske konformasjoner. Legg imidlertid merke til at det er noen rester hvis konformasjoner kartlegger de forbudte områdene. De fleste av disse rester kart i regionen mellom ’tillatt’ regioner 2 og 3, rundt ψ = 0.

-
Se igjen På Figur 4 og forestill deg at du kan vri den øverste peptidgruppen gjennom 180° slik at ψ = 0. Hvilke grupper er sannsynlig å konflikt i denne konformasjonen?
-
N – h-gruppene av tilstøtende peptidgrupper vil komme i konflikt med hverandre, blir tvunget inn i nærhet.
konflikten forbundet med disse konformasjonene kan innkvarteres ved en liten grad av vridning av peptidbindingen. I slike konformasjoner blir peptidgruppen således vridd ut av sin vanlige plan konformasjon.
et begrenset antall ‘forbudte’ konformasjoner av bestemte rester kan tolereres i et polypeptid hvis den vedtatte konformasjonen som helhet er energisk gunstig. Et polypeptid vil ha en tendens til å kaste seg slik at det vedtar den mest stabile konformasjonen. I denne konformasjonen minimerer polypeptidet sin frie energi. I de neste avsnittene skal vi se på dette høyere nivået av proteinstruktur.