- Definisjoner og Prevalens
- Diagnose
- NCCAH Manifestasjoner Hos Kvinner
- Manifestasjoner I Barndommen
- Manifestasjoner I Ungdom og Voksenliv
- NCCAH Ledelse fra Første Manifestasjon Til Voksenlivet
- Behandling I Barndommen
- Behandling I Ungdomsårene
- Pasienter Behandlet Siden Barndommen
- Kvinner Med Første Manifestasjon Under Ungdomsår Eller Hyperandrogen Symptomer etter Seponering av Behandlingen
- NCCAH og Reproduksjon
- Subfertilitet
- Miscarriages
- Fertilitetsplanlegging
- Prenatal Behandling av CAH
- Behandling under Graviditet
- Andre Spesielle Problemer
- Stressmestring I NCCAH
- Psykobiologiske Aspekter i NCCAH
- Konklusjoner
- Forfatterbidrag
- Interessekonflikt
Definisjoner og Prevalens
Congenital adrenal hyperplasia (Cah) omfatter en familie av autosomale recessive lidelser preget av mild til akutt svekket kortisolsyntese på grunn av mangel på ett av de fem adrenal steroidogene enzymene som kreves for kortisolproduksjon (1, 2). Konvensjonelt er CAH delt inn i (a) klassisk (CCAH), presenterer med salt-sløse eller den enkle virilizing form som er manifestert ved fødselen og/eller i nyfødtperioden, og (b) ikke-klassisk (NCCAH), som representerer en mindre alvorlig form for lidelse som mangler genital tvetydighet, er ikke umiddelbart livstruende, og presenterer senere i livet, eller forblir asymptomatisk, eller er feildiagnostisert som en annen sykdom (3). Grensene på tvers AV DE ulike former FOR CAH er imidlertid ikke strengt definert, noe som har en tendens til å øke utfordringene knyttet til denne lidelsen. DET anbefales derfor å vurdere CAH som et kontinuum av fenotyper, fra alvorlig til mild eller asymptomatisk (3).den vanligste formen FOR CAH er forårsaket av 21-hydroksylasemangel (21OHD), noe som resulterer i nedsatt eller ingen konvertering av 17 hydroksyprogesteron (17 OHP) til 11 deoksykortisol og progesteron til deoksykortikosteron. Blokkaden av steroid konvertering resulterer i økt produksjon av androgenforløpere, under CRH-ACTH stimulering, som fører til biokjemisk hyperandrogenisme, preget av forhøyede 17-OHP nivåer. Den klassiske formen av sykdommen, forekommer i 1 av 16.000 levendefødte over hele verden. Det skal imidlertid nevnes at den oppfattede forekomsten av sykdommen er relatert til screeningsmetoden som brukes. For Eksempel har det blitt rapportert at forekomsten av den klassiske formen FOR CAH i Sverige er 1: 11,500 når en case survey-tilnærming brukes, hvilket tallet faller imidlertid til 1:9,800 når hormonell screening brukes. På Samme måte varierer forekomsten I Frankrike og Sveits mellom 1:15.472 og 1:23.000 og 1:10.749 og 1:11.661 når de forskjellige metodene for screening brukes (4).NCCAH er mye hyppigere, forekommer i ca 1 av 1000 Kaukasiere og mer vanlig i visse etniske grupper, slik Som Askenasiske Jøder (1:27), Hispanics (1:53), Jugoslaver (1: 62), Og Italienere (1:300) (5, 6). Likevel mangler data om utbredelsen AV NCCAH basert på ulike estimeringsmetoder (case-survey, hormonell screening og molekylær testing). NCCAH regnes som den vanligste autosomale recessive endokrine lidelsen med en bærefrekvens på 1: 25 til 1:10. I NCCAH på grunn av 21-OHD, er den gjenværende enzymatiske aktiviteten anslått til ca 10-70% (7-9).
Diagnose
i forhold til diagnosen av den klassiske sykdomsformen, som er laget ved fødselen eller i nyfødtperioden på grunn av kjønns tvetydighet og / eller saltutslettende symptomer eller gjennom screeningsprogrammer ansatt i noen land, er de fleste tilfeller AV NCCAH ikke lett detekterbare (4, 10). I tillegg forblir mange individer asymptomatiske i barndommen og ungdomsårene, har normal reproduktiv funksjon, og blir bare oppmerksomme PÅ NCCAH på grunn av diagnosen av et annet familiemedlem og påfølgende testing (11). Imidlertid søker de fleste kvinner med NCCAH medisinsk hjelp når de opplever symptomer på androgenoverskudd, og når klinisk mistanke ber om testing, vil forhøyede basale 17 OHP nivåer mer sannsynlig enn ikke peke på EN diagnose AV NCCAH.faktisk anbefaler de kliniske retningslinjene foreslått av Det Endokrine Samfunnet en baseline ikke-stimulert verdi på 17 OHP som screeningsverktøy for NCCAH. Morgen 17 OHP nivåer >6 nmol / L i follikulærfasen hos menstruerende kvinner bør be om ytterligere evaluering, siden det har vist seg at nivåer over 6 nmol/L fanger 90% AV NCCAH individer (12, 13). Tilfeldige målinger av 17 OHP har ikke vist seg å være nyttige, siden disse ofte gir normale nivåer hos pasienter med NCCAH, og de er dessuten ekstremt høye i lutealfasen hos halvparten av friske kvinner (13). Men i våre data hentet fra 280 personer med sykdommen, seks pasienter (2.1%) hadde en baseline 17 OHP verdi < 6 nmol/L (14). I tillegg Bidet et al., i en stor kohort av kvinner MED NCCAH verifisert av molekylære teknikker også funnet basal 17 OHP verdier lavere enn 6 nmol / L i 8% av forsøkspersonene studert (15). Til slutt, basert på data samlet inn Av Speiser et al. 9% av PERSONER med NCCAH viste 17 OHP-verdier lavere enn 2 ng / ml (som tilsvarer 6nmol/L). Ifølge andre studier er en baseline verdi på 17 OHP mellom 5,1 og 9 nmol/L tilstrekkelig for diagnosen NCCAH (13, 16, 17). Nylig et nivå av basal 17 OHP på 4.6 nmol / L ble foreslått som en terskel FOR ACTH-testing for å forutsi NCCAH hos personer med tidlig adrenarche i barndommen (18).
summen av disse funnene og forslagene indikerer at utvalget av pasienter som skal gjennomgå En Synacthen-stimuleringstest, bør evalueres i hvert enkelt tilfelle. Et 17 OHP poststimuleringsnivå over 3 nmol / L er nødvendig for diagnosen (19). Heterozygoter som bærer EN CYP21A2-mutasjon, viser noe forhøyede 17 OHP-nivåer etter ACTH-stimulering, selv om det er overlapping hos upåvirkede personer (9). Men Som Dacou-Voutetakis et al. har påpekt, hvis summen av basal og post-stimulering 17 OHP verdier overstiger 1,5 nmol / L, så er muligheten for heterozygositet eksepsjonelt høy (20).
Et annet viktig hensyn gjelder teknikkene som brukes til 17 OHP-evaluering. 17 OHP-nivåer måles ved en rekke immunoassay-metoder, men som nylig har vist seg, ble de mest nøyaktige og pålitelige resultatene oppnådd ved implementering av kombinasjonen av væskekromatografi med massespektrometri (LC-MS/MS). Faktisk ble mange falske positive 17 OHP-målinger funnet når lc-MS/Ms-målinger ble sammenlignet med standardmetoder (21) likevel er sistnevnte prosedyrer ikke universelt brukt, i tilfelle en usikker diagnose vil de gi mer presise resultater. Av notatet, screening og diagnostiske terskler FOR LC-MS / MS kvantifisert 17 OHP er ennå ikke definert. Disse terskelene er sannsynligvis lavere enn de som er etablert med immunoassays på grunn av økt spesifisitet AV LC-MS / MS, en metode som er mindre utsatt for kryssreaktivitet og interferenser (22, 23). Hvorvidt en urinsteroidprofil er nødvendig for den endelige diagnosen, gjenstår å bli klarlagt (22). I borderline tilfeller er det tilrådelig å oppnå en komplett adrenokortisk profil etter ACTH-stimuleringstesten for å skille 21-hydroksylasemangel fra andre enzymdefekter og etablere en fast diagnose.spesielt bør en komplett steroidprofil utføres i tvetydige tilfeller for å bekrefte 21-hydroksylasemangel og utelukke andre enzymatiske oppdagelser. Inkludering av 17 OHP, kortisol, 11-deoksykortikosteron,11-deoksykortisol,17-OH-pregnenolon, Dehydroepiandrosteron og androstenedion i en serumsteroid steroidprofil vil være nyttig for å utelukke andre årsaker TIL CAH, som 11β-hydroksylasemangel og, sjeldnere, 3β-hydroksysteroid dehydrogenasemangel Eller P450 oksidoreduktasemangel (24). Mens genetisk testing ikke anses å være et primært diagnostisk verktøy for NCCAH på dette tidspunktet, er det obligatorisk for diagnosebekreftelse og for genetisk rådgivning (3).
når det gjelder kortisolnivåene, forventes det generelt en post-stimuleringsverdi på minst 496 nmol / L. Av notatet, ifølge Stoupa et al. 60% av 47 barn med NCCAH som følge av 21 OHD hadde lave kortisolverdier etter stimuleringen, et funn som peker på behovet for økt overvåking for utvikling av binyreinsuffisiens under store stressorhendelser (25).
NCCAH Manifestasjoner Hos Kvinner
DET kliniske uttrykket FOR NCCAH er preget av et høyt nivå av polymorfisme når det gjelder ikke bare alder av debut, men også de forskjellige tegn og symptomer. DET er rapportert at DEN første kliniske presentasjonen AV NCCAH er i 11% av tilfellene før 10 år og i 80% mellom 10 og 40 år (12). Korrelasjonen mellom genotype og fenotype i CAH og NCCAH er ennå ikke klarlagt. Speiser et al. antyde at de fleste, men ikke alle, fenotypiske variasjoner i 21-hydroksylasemangel skyldes allelisk variasjon I CYP21A2 (26).
Manifestasjoner I Barndommen
I den nyfødte perioden FORBLIR NCCAH-kvinner vanligvis asymptomatiske og har normale ytre kjønnsorganer. DET tidligste tilfellet AV NCCAH rapportert er en 6 måneder gammel jente som utviklet kjønnshår (11). Vanligvis starter kliniske funn og symptomer i NCCAH-tilfeller fra en alder av 5 år eller senere og er relatert til økte androgenivåer, selv om mild kortisolmangel også kan forekomme i noen tilfeller (8). I 92% AV NCCAH-tilfellene er det første symptomet som manifesterer prematur pubarche (PP), som forekommer under 8 år hos jenter og under 9 år hos gutter (12) med en estimert prevalens på mellom 10 og 11,3% (15). AV notatet, hos barn referert MED PP, i henhold til ulike studier, forekomsten AV NCCAH varierer fra 5 til 30% (20, 27). Basert på vår analyse av 280 pasienter med molekylær bekreftelse AV NCCAH, fant vi imidlertid at forekomsten AV PP hos 94 kvinner yngre enn 8 år var så mye som 88%. På den annen side identifiserte en analyse av 45 menn MED NCCAH PP hos bare 29% av pasientene (14). Videre bør vi huske på at omtrent halvparten av personer MED PP kan være heterozygote bærere AV EN CYP21A2-mutasjon.
Et annet aspekt som trenger avklaring er forholdet MELLOM PP og veslevoksen pubertet. Det antas at i noen tilfeller kan perifer aromatisering av adrenal androgener til østrogener aktivere hypotalamus-hypofyse-gonadalaksen, noe som fører til sekundær veslevoksne pubertet (28). I et slikt tilfelle er det nødvendig med regelmessig oppfølging. Noen barn utvikler hirsutisme, akne og / eller rask vekst og manifesterer høy struktur og beinalderutvikling (29). Sistnevnte kan resultere i avkortet slutthøyde som følge av rask epifysisk fusjon. Andre sjeldne kliniske funn i barndommen inkluderer labial adhesjon, perianal hår, klitoromegali og heshet av stemme (30-32).
Manifestasjoner I Ungdom og Voksenliv
UNDER ungdom og voksen alder presenterer NCCAH hos kvinner hyperandrogeniske symptomer som ligner polycystisk ovariesyndrom (PCOS) (33). Symptomer inkluderer hirsutisme, akne, androgen alopecia, anovulasjon, menstrual dysfunksjon og infertilitet. I en multisenterstudie var de vanligste symptomene blant unge og voksne kvinner hirsutisme (59%), oligomenorrhea (54%) og akne (33%). Blant 161 kvinner med NCCAH var symptomer hirsutisme (78%), menstruasjonsdysfunksjon (54,7%) og redusert fruktbarhet (12%) (34). I vår gruppe på 161 kvinner med sykdommen fikk 63% av pasienten polycystisk ovarielignende fenotype (14). Dette funnet har en omvendt implikasjon, siden forekomsten AV NCCAH er høy hos kvinner med hyperandrogenemi. Sikkert, studien Av Witcel et al. rapportering AV EN forekomst AV NCCAH fra 1 til 33% hos hyperandrogene kvinner er av stor interesse. I en annen studie av 950 kvinner som ble henvist til evaluering av androgenoverskudd, BLE NCCAH påvist hos 4,2% av dem. NCCAH-pasientene var imidlertid yngre og mer hirsute sammenlignet med de andre gruppene av hyperandrogene pasienter. De ble også preget av betydelig høyere nivåer av testosteron, fritt testosteron og 17 OHP enn pasienter med andre hyperandrogene syndromer. I ovarial ultralyd viste 77% av dem polycystiske eggstokkene og 41% økte ovariestørrelsen. TIL SAMMEN oppfylte DE PCOS-kriteriene I HENHOLD TIL NIH og Rotterdam med henholdsvis 56 og 72,8% prosent (35).den progressive karakteren av sykdommen er fremhevet av det faktum at forekomsten av hirsutisme har vist seg å øke med alderen og har blitt observert å være sjelden før puberteten. Hirsutisme er den vanligste kliniske manifestasjonen rapportert hos PASIENTER med NCCAH, fra 71 til 96% (28, 36). Pubertale jenter med NCCAH vanligvis tilstede med hirsutisme (37, 38). I den andre enden av spekteret er spørsmålet om alopecia. Mannlig mønster balding er rapportert HOS NCCAH pasienter. Forekomsten av alopecia syntes også å øke med alderen, fra 6% hos pasienter i løpet av det andre tiåret av livet til 19% i femte, noe som igjen indikerer sykdommens progressive karakter (39). Akne er rapportert hos nesten 33% AV NCCAH-fagene (36). Bemerkelsesverdig ble mutasjoner AV CYP21A2-genet som forårsaker NCCAH påvist hos 4,9% av 123 voksne kvinner med alvorlig akne (40).Menstruelle uregelmessigheter inkludert oligomenorrhea eller sekundær amenorrhea kan ofte være presentere tegn PÅ NCCAH i post menarche individer. I tillegg opplever 8-9% av tilfellene også primær amenorrhea, og av denne grunn søker lege for første gang. Videre, BLANT NCCAH individer, oligomenorrhea er mer vanlig enn primær amenorrhea. For eksempel, I en serie Fra Moran et al. blant NCCAH-kvinner i alderen 10 til 19 år var oligomenorrhea tilstede i 56% av tilfellene sammenlignet med bare 9% av ungdomsårene med primær amenorrhea (12). For å konkludere, så mange NCCAH symptomer ligner DE AV PCOS at DET ikke er overraskende AT NCCAH har blitt kalt den store mimicker AV PCOS. I alle fall bør EN DIAGNOSE AV NCCAH vurderes under evalueringen av enhver ung kvinne som henvises til hyperandrogen symptomer.
I en studie Av Arlt et al. PASIENTER med NCCAH ble preget av signifikant høyere BMI og lavere slutthøyde sammenlignet med matchede kontroller (41). Disse dataene, sammen med noen få studier som indikerer tilstedeværelse av insulinsensitivitet, antyder en ugunstig metabolsk profil som kan patofysiologisk forklares av effekten av forhøyede androgenkonsentrasjoner og / eller som følge av glukokortikoidbehandling (42, 43). Osteoporose kan også påvises hos disse personene, sannsynligvis som følge av kortikosteroidbehandlingen (34).
NCCAH Ledelse fra Første Manifestasjon Til Voksenlivet
Når DIAGNOSEN NCCAH er etablert, flere spørsmål knyttet til påfølgende behandlingsanbefalinger berettiger vurdering. Det første spørsmålet som skal tas opp er om glukokortikoidbehandling (GC) er indisert. Den generelle begrunnelsen bak denne tilnærmingen er at ved å gi tilstrekkelig kortisolkonsentrasjoner til pasienten, blir hennes daglige behov dekket, og FØLGELIG VIL CRH-ACTH-aksestimulering bli avsmalnet, noe som fører til redusert adrenal androgenproduksjon. Den generelle regelen om denne tilnærmingen er At GCs gis ved substitusjon og ikke farmakologiske doser, mens påvirkning av alder, kjønn, laboratoriedata, pasientspesifikke anbefalinger og mål for behandling på glukokortikoid erstatningsterapi er alvorlig tatt i betraktning. Vi bør imidlertid også være oppmerksomme på at langvarig gc-substitusjonsbehandling kan føre til hyperkortisolemi, med de resulterende veldokumenterte bivirkningene på alle aspekter av metabolisme, spesielt vekst, fettfordeling og insulinresistens, samt på psykologisk profil. En annen stor ulempe ved denne tilnærmingen er mangelen på en tilstrekkelig klinisk indeks eller biokjemisk markør for tilstrekkelig erstatningsdose, som eksisterer angående TSH-verdier i hypothyroidisme.Til Slutt er det faktum at DET ikke er enighet om hvilken GC SOM skal brukes, og fraværet av langsiktige data om ulike modaliteter AV gc-administrasjon, ytterligere komplisert av behandlende legers beslutninger om og valg av det optimale valget for hver pasient. Hydrokortison brukes vanligvis hos barn, da det ligner mest på det naturlige hormonet (kortisol), men det anses ikke som en egnet tilnærming hos ungdom og unge kvinner på grunn av behovet for flere daglige doser. Derfor foretrekker de fleste voksne endokrinologer enten deksametason eller prednisolon, ved passende doser. På den annen side må det påpekes at ekvivalensen av forskjellige GCs er basert på deres antiinflammatoriske virkning og ikke på forskjellige aspekter av menneskelig metabolisme. Siden kortisol påvirker nesten 20% av det menneskelige genomet, forventes ulike responser av forskjellige GCs i forskjellige vev. Bekrefter denne oppfatningen er det faktum at administrering av deksametason til pasienter MED CAH har vist forverring av indekser av insulinresistens i forhold til andre GCs (44). I tillegg har administrasjonen av 2,5-7,5 mg prednisolon, en dose som anses som normal, en langvarig negativ innvirkning på benmetabolismen (45). Derfor bør hydrokortison anses som den beste behandlingsformen i tilfeller AV gc-tilskuddsbehandling. Fremkomsten av den nylig syntetiserte hydrokortisonformelen med en pille per dag og dens første positive resultater hos pasienter med CAH viser mye løfte for fremtiden (46).
Behandling I Barndommen
NCCAH-pasienter som diagnostiseres i barndommen med TEGN PÅ PP, kan behandles med hydrokortison med sikte på å undertrykke adrenalhormonene og forhindre rask utvikling av beinalder som kan påvirke slutthøyden. Hos de pasientene der hydrokortisonbehandling ble igangsatt 1 år før puberteten og som hadde en beinalder under 9 år, var den endelige høyden innenfor det genetiske potensialet (47). Tilgjengelige studier indikerer at voksen høyde nærmet seg forventet målhøyde hos pasienter som ble nøye overvåket og som strengt overholdt medisineringsplaner (48-50). En nylig liten studie av fem tilfeller (pasienter 6,1–9,2 år) viste at en ultralav dose dexametason er et lovende alternativ for å redusere endogent stress og dets effekter. Hvorvidt dette er en realistisk tilnærming og hvor mye den administrerte dosen skal være gjenstår å bli klarlagt (51). En studie Av Nebesio et al. sammenlignet de hormonelle effektene og farmakokinetikken til hydrokortison, prednisolon og deksametason hos 9 prepubertale pasienter med CCAH. De viste at deksametason var mer potent enn de andre formene for å oppnå betydelig lavere adrenalhormonnivåer, noe som tyder på at deksametason er mer effektivt for undertrykkelse av adrenal androgenproduksjon (46). Selvfølgelig er det nødvendig med videre studier for å verifisere eller avvise dette funnet. Videre kan forhøyede androgenkonsentrasjoner i noen tilfeller føre til sekundær stimulering Av GnRH-aksen, noe som fører til for tidlig pubertet. I et slikt tilfelle kan en parallell kurs med GnRH analog foreskrives dersom beinalderen er signifikant høyere enn kronologisk alder og / eller anslått slutthøyde er uforholdsmessig til målhøyde.
et annet viktig hensyn gjelder tilfeller av samtidig GH-mangel i tilfelle Behandling Med GnRH-analoger og gh-resept resulterte i oppnåelse av målhøyde (52). I henhold til gjeldende retningslinjer anbefales den ovenfor beskrevne terapien forsiktig bare for de tilfellene HVOR den forventede HØYDEN SD er -2,25 < målhøyden eller enda lavere (53). En alternativ indikasjon for oppstart av hydrokortisonbehandling er utilstrekkelig kortisolrespons etter ACTH-stimulering (36).
den foretrukne gc-behandlingen hos barn er vanligvis hydrokortison 10-15 mg / m2, delt inn i tre doser. Ofte har lavere doser også vist seg å være effektive, fra 6 mg / m2 / dag (34). Overdosering bør unngås, vurderer det som kan resultere i dårlig vekst samt Cushingoid funksjoner. Regelmessig vekstmønster, en beinalder som er kompatibel med kronologisk alder, og fravær av sentral fedme kan også fungere som kliniske indekser for riktig ledelse. Når det gjelder biokjemisk/ hormonell profil, betraktes androstenedion og testosteronnivå i midten til øvre områder for beinalder som bedre markører for adekvat gc-erstatningsterapi hos barn med NCCAH. Undertrykte testosteronnivåer ble imidlertid funnet hos 10% AV NCCAH-pasientene, mens ytterligere 28% av pasientene hadde økte testosteronkonsentrasjoner, dette fenomenet kan muligens skyldes hydrokortisonvariabilitet (54, 55). DHEAS-verdiene reduseres med svært små doser, mens undertrykkelsen av 17 OHP og progesteronnivåer krever svært høye gc-doser. Det skal bemerkes at 17 OHP-verdier er avgjørende for diagnosen, men ikke nyttig under oppfølging. Av notatet må blodprøvetaking for hormonell evaluering utføres uten opphør av behandlingen. På grunn av sykdommens forvirring og dens mangesidede natur er det imidlertid ingen spesifikke retningslinjer for tidspunktet for diettendringer eller opphør av glukokortikoidbehandling hos barn.
Behandling I Ungdomsårene
Pasienter Behandlet Siden Barndommen
Til etablering av normal menstruasjon hos NCCAH-jenter, er videreføring Av GCs som startet i barndommen sterkt anbefalt (56). Imidlertid viser ungdomspasienter ofte ikke tilstrekkelig overholdelse av kronisk administrering av legemidler og utelater ofte doser. I puberteten faller også halveringstiden til hydrokortison med 50% som følge av økt IGF-1-nivå, noe som reduserer 11ßohsd-aktivitet, samt på grunn av økt kortisolclearance som følge av forsterkning av glomerulær filtrasjonshastighet (57). Hos den unge kvinnen, 2 år etter menarche, og hvis normale ovulatoriske sykluser er registrert, anbefales en pasient-sentrert tilnærming mot de hyperandrogeniske symptomene som kan oppstå.
hvis det er alvorlige hyperandrogeniske funn, som hirsutisme, akne og/eller oligomenorrhea, vil fortsatt behandling vurderes. Det er viktig å huske de skjøre følelsene til en ung ungdom, som vil bli alvorlig skadet av «avstøtende» tegn på hyperandrogenisme. Kombinasjonen av hyperandrogenic tegn, menstruasjonsforstyrrelser, og dårlig livskvalitet er godt dokumentert, og det er spesielt høy i yngre kvinner. Et annet punkt som skal vurderes er responsen av adrenal reserve post ACTH stimulering. Gitt at toppkortisol av pubertale og voksne kvinner etter stimulering er under 496 nmol / L, i tilfeller av stress, bør steroidbehandling administreres (58). På den annen side, hvis ingen av symptomene ovenfor oppstår hos den unge pasienten, KAN gc-behandlingen avbrytes, hvoretter regelmessig oppfølging anbefales.
Kvinner Med Første Manifestasjon Under Ungdomsår Eller Hyperandrogen Symptomer etter Seponering av Behandlingen
i nærvær av hyperandrogen symptomer anbefales en pasientorientert tilnærming sterkt, med fokus på pasientens hovedklager. Dette er langt bedre enn en generell universell modus operandi, siden den unge pasienten med sistnevnte tilnærming vil bli skuffet, dette fører til seponering av behandlingen og som følge av uønskede helseutfall i voksen alder. I tilfeller med alvorlig hirsutisme utgjør et kurs på 6 til 12 måneder med orale prevensjonspiller (OCPs) en rimelig prosedyre gitt de gunstige effektene Av OCPs på shbg leverproduksjon og reduksjon av androgenfrigivelse fra eggstokken. Klinisk bedring forventes etter Minst 2-3 måneders Oppstart Av OCPs, mens samtidig bruk av et progestagen med minimale androgene egenskaper er sterkt anbefalt. Hvis OCPs mislykkes som førstelinjetilnærming, kan antiandrogener (spironolakton, flutamid, bikalutamid, cyproteronacetat og finasterid) tilsettes til behandlingen. Etter vår erfaring har administrasjonen av bikalutamid oppnådd betydelig forbedring i tilfeller av alvorlig akne, men lignende resultater ble ikke oppnådd i tilfeller med alvorlig hirsutisme. Kosmetiske tilnærminger som laserapplikasjon og depilatories kan også foreslås for kvinner som klager over overdreven eller uønsket hårvekst eller hodebunnshårtap (androgen alopecia) (32, 34, 57).
hos pasienter med utilstrekkelig kortisolsekresjon etter stimulering, eller Hvis OCPs og antiandrogener ikke kan tolereres, anbefales et kurs med GCs sterkt (57). Data Fra New et al. indikerer at uregelmessig menstruasjon og akne reverseres innen 3 måneder etter oppstart av glukokortikoidbehandling, mens hirsutisme krever nesten 30 måneder (59). I motsetning til barndommen, i ungdomsår, brukes ofte langtidsvirkende steroider, og regimer på 5 mg prednisolon eller 0,25 mg dexametason anbefales (53). I den virkelige verden har imidlertid kliniske data vist en rekke forskjellige regimer brukt I NCCAH-ledelse (41).
det primære målet med behandlingen bør være pasientens lindring fra de hyperandrogeniske symptomene. Blant personer med NCCAH diagnostisert utelukkende basert på laboratorieresultater, bør kliniske egenskaper derfor veilede anbefalinger fra ledelsen, siden hormonelle og molekylære funn ikke nødvendigvis forutsier klinisk alvorlighetsgrad. I Henhold Til Endocrine Society-retningslinjene bør NCCAH-pasienter få muligheten til å avbryte gc-behandling når symptomene går over (60). Selvfølgelig bør disse pasientene ikke gå tapt for oppfølging, mens behandlingen bør gjenopptas ved tilbakefall av symptomene. Videre, ved seponering, bør pasientene informeres om muligheten for infertilitet og bør oppfordres til å søke lege hvis de ønsker å bli gravide (19). Av notatet, bør riktig overføring av pasienten fra pediatrisk til voksen endokrinolog utføres, optimalt etter 1 år med synkronisert overvåking(53, 61).
NCCAH og Reproduksjon
Subfertilitet
Subfertilitet er vanlig rapportert i klassiske former FOR CAH, som hovedsakelig skyldes menstruasjonsforstyrrelser, kronisk anovulasjon og anatomiske deformiteter. Fødselsraten er anslått til 17% i forhold til 65% av kontrollpopulasjonen (62). I mellomtiden har data om fruktbarhet hos kvinner med NCCAH nylig blitt vurdert i detalj, og den estimerte infertilitetsforekomsten er 11% blant NCCAH-kvinner, det vil si relativt mildere enn I CAH, og i mange tilfeller er det lett å løse. Bidet et al. evaluert fruktbarhet hos 190 kvinner som lider AV NCCAH, hvorav 95 ønsket å bli gravid. I denne populasjonen ble det rapportert 187 graviditeter hos 85 kvinner, noe som resulterte i 141 fødsler hos 82 personer. Det må fremheves at 99 av svangerskapene (52,9%) skjedde før DIAGNOSEN NCCAH, tre av dem med anvendelse av eggløsningsinduksjonsprotokoller, resten var spontane, mens 88 graviditeter fant sted etter NCCAH-diagnose. I de aller fleste av dem utviklet graviditeten etter institusjonen av terapi med hydrokortison, mens hos 11 kvinner skjedde det spontant (63, 64). I en annen rapport av 22 observerte NCCAH kvinner som ønsker graviditet, fulgte 12 graviditeter med prednison (65).
Miscarriages
i kohorten Av Bidet et al. abortraten var henholdsvis 6,5 og 26,3% hos pasienter behandlet med GCs og ubehandlede pasienter (64). Utfallet av graviditeten kan til og med lykkes uten glukokortikoidbehandling i tilfeller DER NCCAH ennå ikke ble diagnostisert, som også rapportert I en saksrapport Fra Cuhaci et al. Likevel opplevde det samme paret to miscarriages og rapporterte subfertilitet etter denne første graviditeten (66). Så langt som muligheten for prematur graviditet er bekymret, som vurdert Av Bidet et al., det skiller seg ikke vesentlig fra resten av befolkningen (15).
Fertilitetsplanlegging
FOR de kvinnene med symptomatisk hyperandrogenisme eller med rapportert infertilitet, men som ønsker å bli gravide, ANBEFALES GC-behandling sterkt. I løpet av denne perioden er glukokortikoider som brukes prednison eller hydrokortison. Den viktigste handlingen, fordi den vil lede de påfølgende trinnene, er genetisk testing av de potensielle foreldrene. Kvinner MED NCCAH som ønsker å bli gravid, bør være oppmerksomme på risikoen for å føde et spedbarn med den klassiske sykdomsformen. Utfallet av svangerskap blant kvinner med NCCAH, og mer spesifikt forekomsten av spedbarn født med CCAH, er estimert i nyere studier til å være mellom 1,5 og 2,5%, MED NCCAH på ca 15% (36, 64). Selvfølgelig avhenger denne frekvensen av referansepopulasjonen, en høyere forekomst som forekommer i populasjoner med høye inngifte priser (36). De forventede sjansene for at foreldre får BARN med CAH er 1 av 120 for ukjent fadergenotype, null hvis faren ikke har relevant mutasjon, og 1 av 4 hvis han har en heterozygot mutasjon (19). Som et resultat er genetisk testing av partnerne til disse kvinnene viktig for å vurdere risikoen for å føde et barn med den klassiske formen AV CAH (64, 67).
i tilfelle at den potensielle moren bærer to nc-mutasjoner (Tabell 1), er det ikke absolutt behov for genetisk testing i fremtiden far, siden han er heterozygot FOR CYP12A2-mutasjonene, er avkomene i fare for å utvikle den ikke-klassiske sykdomsformen i fremtiden. Det skal imidlertid bemerkes at bærestatus er 2-15% i forskjellige populasjoner, og halvparten av disse personene har en alvorlig mutasjon. Videre er definitive anbefalinger om situasjoner der genetisk testing ikke er nødvendig vanskelig gitt den ufullkomne genotype-fenotype korrelasjonen, spesielt for mildere mutasjoner . FOR eksempel FOREKOMMER p30l-mutasjonen, oftest assosiert MED NCCAH, også hos pasienter med klassisk CAH . En stor europeisk multisenterstudie viste nylig at dette var tilfelle hos 78% av pasientene . I denne studien var den milde v218l-mutasjonen assosiert Med Klassisk snarere ENN NCCAH i 30% av tilfellene. DERFOR bør alle pasienter med NCCAH tilbys genetisk rådgivning og molekylær vurdering av reproduktive partnere.
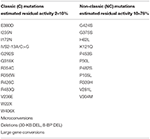
Tabell 1. Mutasjoner AV CYP21A2 med minimal (C) eller moderat (nc) rest enzymaktivitet (14, 68, 69).i kontrast, i tilfelle at den potensielle moren er en sammensatt heterozygot med en alvorlig (C) og EN nc-mutasjon, er genetisk testing av den fremtidige far obligatorisk. Vi bør huske på at data som stammer fra studier som bruker immunoassays eller de mer nøyaktige lc-MS/MS-prosedyrene, konsekvent fremhever manglende evne til pålitelig å utelukke heterozygositet ved bruk av basal-eller ACTH-stimulerte 17 OHP-verdier, på grunn av den signifikante overlappingen med det normale området. Man kan foreslå bruk Av En Synacthen-test, og hvis DEN ikke er kompatibel med heterozygositet (summen av basal og toppstimulert 17 OHP-verdier < 1,5 nmol/L), KAN DNA-testing unngås. Vi bør imidlertid være svært forsiktige i tolkningen av denne testen, siden den ikke er validert i andre studier. Derfor kan BRUK AV ACTH stimulert 21-deoksykortisol enten enkeltvis eller som en del av et steroidforhold eller steroidprofil, lette den biokjemiske identifikasjonen av heterozygoter i fremtiden, særlig ETTERSOM LC-MS/MS blir mer tilgjengelig . Hvis den potensielle far bære EN NC mutasjon, så ingenting annet er nødvendig. Omvendt, hvis han er bærer Av En Klassisk mutasjon, vil spørsmålet om prenatal behandling av fosteret oppstå. Alle mulige genotypekombinasjoner og de foreslåtte prosedyrene er presentert I Tabell 2.

Tabell 2. Planlegging av foreslåtte prosedyrer under graviditet basert på potensielle foreldre genotype.
Prenatal Behandling av CAH
målet med prenatal behandling av cah er forebygging av genital virilisering av fosteret, men også lindring av stress følte av foreldrene som er sannsynlig å ha et barn med tvetydige genitalia (70). Deksametason brukes på grunn av sin evne til å krysse placenta og fordi den ikke er deaktivert fra placenta 11β OH steroid dehydrogenase og bindes kun minimalt til mors blod kortisolbindende globulin (CBG). Deksametason, ved å undertrykke fetal acth sekresjon, reduserer forhøyet fetal androgen produksjon. Behandlingen må avbrytes dersom karyotype eller DNA-analyse viser henholdsvis en hann eller en upåvirket hunn (71). Likevel, selv om føtal genital virilisering allerede har startet på 6-7 uker etter unnfangelsen, kan chorioniske villøse biopsier for genetisk diagnose ikke oppnås før 10. -12. uke. Dette tidsintervallet antyder at alle graviditeter med risiko for virilisering AV CAH bør behandles, selv om bare 1 av 8 fostre er berørt og kvinner (19, 34, 70).
hvorvidt denne eksponeringen av fosteret for deksametason for forebyggende tiltak er medisinsk og etisk akseptabelt forblir kontroversielt (70). Dyrestudier har vist at deksametason i første trimester reduserer fødselsvekt, påvirker betacellefunksjonen i nyrene i bukspyttkjertelen og hjernens utvikling, og predisponerer for angst, hypertensjon og hyperglykemi i voksen alder (70). Videre klager 10-20% av gravide kvinner som bruker dexamethasonbehandling under graviditet på vektøkning, økt appetitt, humørsvingninger, søvnløshet, edemas, hypertensjon, hyperglykemi og striae. Men New et al. fant en lavere Enn gjennomsnittlig Prader-score hos fostre behandlet med deksametason prenatalt, men ingen forskjell i langsiktige utfall (72). I tillegg, I en gjennomgang Av Merce Fernandez-Balsells et al. deksametason ble vist å være assosiert med reduksjon i fostervirilisering uten signifikante bivirkninger hos mor eller foster (63). Forfatterne av ovennevnte gjennomgang samt de påfølgende retningslinjene For Det Endokrine Samfunnet påpeker imidlertid at på grunn av de små utvalgsstørrelsene i hele litteraturens kropp, er emnet usikkert og det er klart behov for ytterligere undersøkelser.beslutningen om initiering av behandling bør kun foretas i store sentre med et erfarent team og protokoller godkjent Av Institusjonelle Kontrollgrupper og basert på familiens verdier og preferanser og med skriftlig informert samtykke som en forutsetning (19, 63). Flere spesialister på feltet indikerer at en høyere verdi bør plasseres på å hindre unødvendig prenatal eksponering av mor og foster til deksametason i stedet for å pålegge den emosjonelle toll av tvetydige genitalia på foreldre og pasienter. Viktigst, det bør avklares for foreldre at deksametasonadministrasjon ikke endrer pasientstatus, men er rettet mot å redusere behovet for kirurgi i stedet for å bevare liv eller intellektuell kapasitet. De må også vite at sannsynligheten for at deres barn vil lide av den klassiske sykdomsformen er høy, til tross for behandling. Mest avgjørende, i mellomtiden, eksponeringen av en ung organisme til en meget potent GC i en spesielt følsom periode med fosterprogrammering og vekst, som godt kan vise seg ubrukelig i tilfelle av et foster MED xy karyotype, støttes for tiden ikke av robuste og ubestridelige data.Ideelt sett bør det være en tidlig diagnose, dette utføres via en ikke-invasiv teknikk og før begynnelsen av kjønnsvirilisering av fosteret. Arbeid i denne retningen peker nye studier på bruken av cellefritt føtal DNA oppnådd fra mors plasma som en lovende metode som vil tillate bestemmelse av føtal kjønn og diagnose AV CAH i tidlig svangerskapsalder (<9 uker). Hvis denne prosedyren er omfattende implementert i klinisk praksis, vil unødvendig prenatal deksametasonbehandling unngås (73).
Behandling under Graviditet
under graviditet bør kvinner med NCCAH fortrinnsvis behandles med hydrokortison, ikke bare fordi graviditet vanligvis ledsages av forhøyede stressnivåer, men også som et forebyggende tiltak mot den ovennevnte høye forekomsten av miskramper. Denne formen for glukokortikoid er foretrukket på grunn av dets metabolisering av placentaenzym 11β OH steroid dehydrogenase II, som forhindrer glukokortikoid fra å ha noen effekt på fosteret (71). Ved unnfangelsen bør hydrokortisondosen økes til 20-25 mg / dag, og dosejusteringer utføres hver 6-8 uker med sikte på å holde testosteronnivåene i de øvre normale nivåene av graviditetstraffen (74). Som et eksempel vises testosteron-og D4-verdiene til en pasient MED NCCAH fra unnfangelse opptil 3 år etter levering av to friske tvillinger (Figur 1).
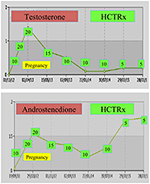
Figur 1. Testosteron-og andostenedionnivåer og påfølgende hydrokortison-dosering (HCTRx), fra unnfangelse til 3 år etter levering administrert til en pasient MED NCCAH.
Andre Spesielle Problemer
Stressmestring I NCCAH
under alvorlig livstruende stress, kirurgi eller alvorlig sykdom, kan PASIENTER med NCCAH som behandles med glukokortikoider som behandles med glukokortikoider, kreve større eller hyppigere doser fordi binyrefunksjonen er iatrogen undertrykt. Det er derfor viktig å utdanne foreldrene til små barn, samt å re-utdanne pasienter på deres overgang til voksen omsorg, om stress dosering. FOR NCCAH-pasienter som ikke behandles Med GCs eller ved seponering, bør kortisolrespons på ACTH vurderes. Nesten en tredjedel AV NCCAH-pasientene reagerer utilstrekkelig på stimuleringen (15, 75). For de som responderer NORMALT PÅ acth-stimulering anbefales ingen behandling med stressdoser (19). Mineralokortikoidbehandling er ikke nødvendig i noen av tilfellene, da disse pasientene har normal aldosteronsekresjon og ikke utvikler saltavfall (58). I tilfelle av hypertyreose eller en økning i levotyroksinbehandling, øker kortisolclearance og en binyrekrise kan oppstå (58, 76).
Psykobiologiske Aspekter i NCCAH
Meyer-Bahlburg et al. har i flere studier rapportert nedsatt psykologisk profil blant pasienter med NCCAH på GRUNN AV 21oh mangel. Mer spesifikt hadde de berørte kvinnene en høyere masculinization / defeminization score på flere tiltak av kjønnsrelatert atferd sammenlignet med normal kontroll kvinner, men markert mindre enn hos kvinner med klassisk CAH. De hadde også betydelig økt biseksuell eller homoseksuell orientering. Retrospektive kliniske kvalitative intervjuer med disse kvinnene viste en historie med ubehag og sosialt stress relatert til deres pre-behandling erfaringer med androgenavhengige tegn, som akne, hirsutisme og unnfangelsesvansker (59, 77, 78). Tilsvarende, Arlt et al. viste at subjektiv helsestatus var signifikant svekket på tvers av alle åtte domener i en kortsiktig helseundersøkelse, med de mest fremtredende forskjellene, sammenlignet med alder – og kjønnsmatchede kontroller, knyttet til domenene generell helse, vitalitet og rollebegrensninger på grunn av følelsesmessige problemer. Spørreskjemaet Hospital Anxiety And Depression Scale (HADS) viste også økte angstpoeng (41). Med tanke på disse funnene bør psykologiske parametere for å veilede terapi vurderes hos kvinner med NCCAH, og i sammenheng med pasientorientert tilnærming må en psykologisk diagnose og støtte tilbys.
Konklusjoner
NCCAH anses som hyppigere og mildere form AV CAH på grunn av retensjon av 20-50% enzymaktivitet. De fleste pasienter kan søke lege når som helst i livet på grunn av symptomer relatert til androgenoverskudd. Disse inkluderer for tidlig pubarche, rask vekst, hirsutisme, akne, menstruelle uregelmessigheter eller infertilitet, samt mange andre mindre fremtredende manifestasjoner. Tidlig morgen baseline verdier av 17 OHP som en god første screening test og videre evaluering MED ACTH stimulering og, i tilfelle av borderline resultater, genetisk testing, anbefales. Som en generell regel bør androstenedion og ikke 17 OHP-nivåer brukes under oppfølging.
behandlingsbeslutningen bør baseres på vurdering av fakta og bør følge en individualisert tilnærming. Behandling er ikke alltid indisert med mindre pasienten er symptomatisk, for eksempel barn med tidlig debut og rask progresjon av kjønnshår og kroppshår, rask vekst, og / eller skjelettutvikling, eller kvinner med oligomenorrhea, akne, hirsutisme, infertilitet, eller en kombinasjon av disse og andre av de ovennevnte symptomer. Genetisk rådgivning er sterkt anbefalt I NCCAH kvinner som ønsker å bli gravide, samt genotyping av far. Den generelle behandlingen av pasienten inkluderer i tillegg behandling av sannsynlige komplikasjoner av glukokortikoidbehandling eller metabolismerelaterte manifestasjoner av sykdommen. Til slutt, gitt at mange terapeutiske problemer knyttet til riktig behandling av disse pasientene ennå ikke er klarlagt, er det svært viktig for den behandlende legen å holde seg oppdatert med all utvikling på dette feltet og å integrere de nye dataene i sin kliniske praksis. Sikkert, ytterligere kliniske studier på dette området er avgjørende.FOR Å oppsummere, FOR NCCAH-kvinnen, er den ideelle tilnærmingen en skreddersydd, og innlemmer en jevn overgang av hennes ledelse når hun er henvist fra pediatrisk til voksen endokrinolog, sammen med symptomorientert behandling som vil følge henne gjennom hele livet. Gitt de mange faktorene som påvirker det hyperandrogeniske systemet, er det tilrådelig å oppmuntre pasienter til å vedta en sunnere livsstil ved å forbedre kostvaner, øke trening og sikte på vektreduksjon. Videre er psykologisk støtte i visse tilfeller ofte gunstig. Videre spesialister i felt som er involvert i behandling av denne lidelsen, som hudleger, gynekologer og psykologer, trenger inngående forståelse om håndtering av mistenkelige eller allerede diagnostisert tilfeller AV NCCAH. For å konkludere, la oss huske det innsiktsfulle sitatet Fra Den Amerikanske astronomen Vera Cooper Rubin: «Vitenskapen utvikler seg best når observasjoner tvinger oss til å endre våre forutsetninger.»
Forfatterbidrag
alle forfattere oppført har gjort et betydelig, direkte og intellektuelt bidrag til arbeidet, og godkjent det for publisering.
Interessekonflikt
forfatterne erklærer at forskningen ble utført i fravær av kommersielle eller økonomiske forhold som kan tolkes som en potensiell interessekonflikt.
1. Speiser PW, Hvit PC. Medfødt adrenal hyperplasi. N Engl J Med. (2003) 349:776–88. doi: 10.1056 / NEJMra021561
PubMed Abstrakt | CrossRef Fulltekst/Google Scholar
2. Turcu AF, Auchus RJ. Adrenal steroidogenesis and congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. (2015) 44:275–96. doi: 10.1016/j.ecl.2015.02.002
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Speiser PW. Nonclassic adrenal hyperplasia. Rev Endocr Metab Disord. (2009) 10:77–82. doi: 10.1007/s11154-008-9097-x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Gidl hryvnas S, Falhammar H, Thilhammar H, von D@beln U, Ritzé M, Wedell A, et al. Ett hundre år med medfødt adrenal hyperplasi I Sverige: en retrospektiv, populasjonsbasert kohortstudie. Lancet Diabetes Endocrinol. (2013) 1:35–42. doi: 10.1016/S2213-8587(13)70007-x
PubMed Abstrakt | CrossRef Fulltekst | Google Scholar
5. Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, New MI. Høy frekvens av ikke-klassisk steroid 21-hydroksylasemangel. Am J Hum Genet. (1985) 37:650–67.
PubMed Abstract | Google Scholar
6. Wilson RC, Nimkarn S, Dumic M, Obeid J, Azar MR, Azar M, et al. Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Mol Genet Metab. (2007) 90:414–21. doi: 10.1016/j.ymgme.2006.12.005
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Miller WL. Clinical review 54: genetics, diagnosis, and management of 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1994) 78:241–6. doi: 10.1210/jcem.78.2.8106606
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Witchel SF, Azziz R. Nonclassic congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2010) 2010:625105. doi: 10.1186/1687-9856-2010-625105
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Wilson RC, Mercado AB, Cheng KC, NEW MI. Steroid 21-hydroksylase mangel: genotype kan ikke forutsi fenotype. J Clin Endocrinol Metab. (1995) 80:2322–9. doi: 10.1210 / jc.80.8.2322
CrossRef Fulltekst/Google Scholar
10. Hvit PC. Neonatal screening for medfødt adrenal hyperplasi. Nat Rev Endocrinol. (2009) 5:490–8. doi: 10.1038 / nrendo.2009.148
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Kohn B, Levine LS, Pollack MS, Pang S, Lorenzen F, Levy D, et al. Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. (1982) 55:817–27. doi: 10.1210/jcem-55-5-817
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Moran C, Azziz R, Carmina E, Dewailly D, Fruzzetti F, Ibañ L, Et al. 21-Hydroksylase-mangelfull ikke-klassisk adrenal hyperplasi er en progressiv lidelse: en multisenterstudie. Am J Fødselslege Gynecol. (2000) 183:1468–74. doi: 10.1067 / mob.2000.108020
PubMed Abstrakt | CrossRef Fulltekst/Google Scholar
13. Azziz R, Hincapie LA, Knochenhauer ES, Dewailly D, Fox L, Støvler Lr. Screening for 21-hydroksylase-mangelfull ikke-klassisk adrenal hyperplasi blant hyperandrogen kvinner: en prospektiv studie. Fertil Steril. (1999) 72:915–25. doi: 10.1016/S0015-0282(99)00383-0
PubMed Abstrakt | CrossRef Full Tekst | Google Scholar
14. A-M, A-M, A-M, A-M, a-M, a-M, a-M, a-M, a-M, a-M, a-M, a-m, a-m, a-m, a-m, a-m, a-m, a-m, a-m, a-m, et al. Spekteret av kliniske, hormonelle og molekylære funn hos 280 personer med ikke-klassisk medfødt adrenal hyperplasi forårsaket av MUTASJONER AV CYP21A2-genet. Clin Endocrinol (Oxf). (2015) 82:543–9. doi: 10.1111 / cen.12543
PubMed Abstrakt | CrossRef Fulltekst / Google Scholar
15. Bidet M, Bellann ④ Chantelot C, Galand-Portier M-B, Tardy V, Billaud L, Laborde K, et al. Klinisk og molekylær karakterisering av en kohorte av 161 urelaterte kvinner med ikke-klassisk medfødt adrenal hyperplasi på grunn av 21-hydroksylasemangel og 330 familiemedlemmer. J Clin Endocrinol Metab. (2009) 94:1570–8. doi: 10.1210 / jc.2008-1582
PubMed Abstrakt | CrossRef Fulltekst/Google Scholar
16. Escobar-Morreale HF, Sanch Hryvn R, San Millá Jl. En prospektiv studie av forekomsten av ikke-klassisk medfødt adrenal hyperplasi blant kvinner med hyperandrogen symptomer og tegn. J Clin Endocrinol Metab. (2008) 93:527–33. doi: 10.1210 / jc.2007-2053
PubMed Abstrakt | CrossRef Fulltekst/Google Scholar
17. Dewailly D, Vantyghem-Haudiquet MC, Sainsard C, Buvat J, Cappoen JP, Ardaens K, et al. Kliniske og biologiske fenotyper ved sen 21-hydroksylasemangel. J Clin Endocrinol Metab. (1986) 63:418–23. doi: 10.1210 / jcem-63-2-418
PubMed Abstrakt | CrossRef Fulltekst/Google Scholar
18. Gö hryvnja EN, Oz Hryvnja ZA, Alikaş I En, Engiz O, Bulum B, Kandemir N. (2011). Er basal serum 17-OH progesteron en pålitelig parameter for å forutsi ikke-klassisk medfødt adrenal hyperplasi i tidlig adrenarche? Turk J Pediatr. 53:274–80.
PubMed Abstrakt / Google Scholar
19. Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, et al. Medfødt adrenal hyperplasi på grunn av steroid 21-hydroksylase mangel: en endocrine society klinisk praksis retningslinje. J Clin Endocrinol Metab. (2010) 95:4133–60. doi: 10.1210 / jc.2009-2631
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Dacou-Voutetakis C, Dracopoulou M. High incidence of molecular defects of the CYP21 gene in patients with premature adrenarche. J Clin Endocrinol Metab. (1999) 84:1570–4. doi: 10.1210/jc.84.5.1570
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Ambroziak U, Kepczynska-Arrangementer i, Kurylowicz, Ma ④unowicz EM, Wó, Miskiewicz P, et al. Diagnosen av ikke-klassisk medfødt hyperplasi av binyrene på grunn av 21-hydroksylasemangel, basert på serumbasal eller post-ACTH stimulering 17-hydroksyprogesteron, kan føre til falsk positiv diagnose. Clin Endocrinol (Oxf). (2016) 84:23–9. doi: 10.1111 / pris 12935
PubMed Abstrakt | CrossRef Fulltekst/Google Scholar
22. Ambroziak Y, Kepczynska-Arrangementer i, Kurylowicz,Wysłouch-Cieszyń A, Mał EM, Bartoszewicz, Et al. LC-MS / MS forbedrer screening mot 21-hydroksylase mangel. Gynecol Endocrinol. (2015) 31:296–300. doi: 10.3109/09513590.2014.994599
PubMed Abstrakt | CrossRef Full Tekst | Google Scholar
23. bbc, CC, Goldman MM, Zhang K, Clark-N, Reitz RE Welt CK. Samtidig måling av tretten steroidhormoner hos kvinner med polycystisk ovariesyndrom og kontroll kvinner ved hjelp av væskekromatografi-tandem massespektrometri. PLoS One. (2014) 9: e93805. doi: 10.1371 / tidsskrift.pone.0093805
PubMed Abstrakt | CrossRef Fulltekst/Google Scholar
24. Ray JA, KUSHNIR MM, Yost RA, Rockwood AL, Wayne Meikle A. ytelsesforbedring i måling av 5 endogene steroider VED LC-MS / MS kombinert med differensial ion mobilitet spektrometri. Clin Chim Acta. (2015) 438:330–6. doi: 10.1016 / j. cca.2014.07.036
PubMed Abstrakt | CrossRef Fulltekst/Google Scholar
25. Stoupa A, Gonz@lez-Briceñ L, Pinto G, Samara-Boustani D, Thalassinos C, Flechtner I, et al. Utilstrekkelig kortisolrespons på tetracosactide (Synacthen®) – testen ved ikke-klassisk medfødt adrenal hyperplasi: et unntak fra regelen? Horm Res Paediatr. (2015) 83:262–7. doi: 10.1159/000369901
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. (1992) 90:584–95. doi: 10.1172/JCI115897
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Temeck JW, Pang SY, Nelson C, New MI. Genetiske defekter av steroidogenese i tidlig pubarche. J Clin Endocrinol Metab. (1987) 64:609–17. doi: 10.1210 / jcem-64-3-609
PubMed Abstrakt | CrossRef Fulltekst/Google Scholar
28. Voutilainen R, J ④elä J. Prematur Adrenarche: etiologi, kliniske funn og konsekvenser. J Steroid Biochem Mol Biol. (2015) 145:226–36. doi: 10.1016/j. jsbmb.2014.06.004
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Oberfield SE, Sopher AB, Gerken AT. Approach to the girl with early onset of pubic hair. J Clin Endocrinol Metab. (2011) 96:1610–22. doi: 10.1210/jc.2011-0225
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Janus D, Wojcik M, Malunowicz E, Starzyk JB. Et tilfelle av tilbakevendende labial adhesjon i et 15 måneder gammelt barn med asymptomatisk ikke-klassisk medfødt adrenal hyperplasi på grunn av 21-hydroksylasemangel. J Pediatr Endocrinol Metab. (2012) 25:1017–21. doi: 10.1515 / jpem-2012-0190
PubMed Abstrakt | CrossRef Fulltekst / Google Scholar
31. Gopal-Kothandapani JS, Petkar A, O ‘ Shea E, Banerjee I. Perianal hår Som en uvanlig presentasjon av ikke-klassisk medfødt adrenal hyperplasi. BMJ Tilfelle Rep. (2013) 2013: bcr2013010123. doi: 10.1136/bcr-2013-010123
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Falhammar H, Nordenström A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine. (2015) 50:32–50. doi: 10.1007/s12020-015-0656-0
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Chrousos GP, Loriaux DL, Mann DL, Cutler GB. Late-onset 21-hydroxylase deficiency mimicking idiopathic hirsutism or polycystic ovarian disease. Ann Intern Med. (1982) 96:143–8. doi: 10.7326/0003-4819-96-2-143
CrossRef Full Text | Google Scholar
34. Witchel SF. Non-classic congenital adrenal hyperplasia. Steroids. (2013) 78:747–50. doi: 10.1016/j.steroids.2013.04.010
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Carmina E, Rosato F, Jannì A, Rizzo M, Longo RA. Extensive clinical experience: relative prevalence of different androgen excess disorders in 950 women referred because of clinical hyperandrogenism. J Clin Endocrinol Metab. (2006) 91:2–6. doi: 10.1210/jc.2005-1457
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Moran C, Azziz R, Weintrob N, Witchel SF, Rohmer V, Dewailly D, et al. Reproduktivt utfall av kvinner med 21-hydroksylase-mangelfull ikke-klassisk adrenal hyperplasi. J Clin Endocrinol Metab. (2006) 91:3451–6. doi: 10.1210 / jc.2006-0062
PubMed Abstrakt | CrossRef Fulltekst/Google Scholar
37. Levin JH, Carmina E, Lobo RA. Er den upassende gonadotropinsekresjonen hos pasienter med polycystisk ovariesyndrom lik den hos pasienter med medfødt adrenal hyperplasi hos voksne? Fertil Steril. (1991) 56:635–40. doi: 10.1016/S0015-0282(16)54592-0
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Carmina E, Lobo RA. Ovarian suppression reduces clinical and endocrine expression of late-onset congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Fertil Steril. (1994) 62:738–43. doi: 10.1016/S0015-0282(16)56998-2
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Ludwig E. Klassifisering av typer androgenetic alopecia (vanlig skallethet) som forekommer hos det kvinnelige kjønn. Br J Dermatol. (1977) 97:247–54. doi: 10.1111/j.1365-2133.1977.tb15179.x
PubMed Abstrakt | CrossRef Fulltekst / Google Scholar
40. A, a, A, A, A, A, A, A, A, A, a, a, a, a, a, et al. Prevalens av ikke-klassisk medfødt adrenal hyperplasi på grunn av 21-hydroksylasemangel hos greske kvinner med akne: en sykehusbasert tverrsnittsstudie. J Eur Acad Dermatol Venereol. (2013) 27:1448–51. doi: 10.1111/j.1468-3083. 2012. 04613.x
PubMed Abstrakt | CrossRef Fulltekst / Google Scholar
41. Arlt W, Willis DS, Wild SH, Krone N, Doherty EJ, Hahner S, et al. Helsestatus hos voksne med medfødt adrenal hyperplasi: en kohortstudie av 203 pasienter. J Clin Endocrinol Metab. (2010) 95:5110–21. doi: 10.1210 / jc.2010-0917
PubMed Abstrakt | CrossRef Fulltekst/Google Scholar
42. Saygili F, Oge A, Yilmaz C. Hyperinsulinemi Og insulin ufølsomhet hos kvinner med ikke-klassisk medfødt adrenal hyperplasi på grunn av 21-hydroksylase mangel: forholdet mellom serum leptinnivåer og kronisk hyperinsulinemi. Horm Res. (2005) 63: 270-4. doi: 10.1159/000086363
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Speiser PW, Serrat J, New MI, Gertner JM. Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1992) 75:1421–4. doi: 10.1210/jc.75.6.1421
PubMed Abstract | CrossRef Full Text | Google Scholar
44. Han TS, Stimson RH, Rees DA, Krone N, Willis DS, Conway GS, et al. Glukokortikoidbehandlingsregime og helseutfall hos voksne med medfødt adrenal hyperplasi. Clin Endocrinol. (2013) 78:197–203. doi: 10.1111 / cen.12045
PubMed Abstrakt | CrossRef Fulltekst / Google Scholar
45. Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Bruk av orale kortikosteroider og risiko for brudd. J Bein Miner Res. (2000) 15: 993-1000. doi: 10.1359 / jbmr.2000.15.6.993
PubMed Abstract | CrossRef Full Text | Google Scholar
46. Nebesio TD, Renbarger JL, Nabhan ZM, Ross SE, Slaven JE, Li L, et al. Differential effects of hydrocortisone, prednisone, and dexamethasone on hormonal and pharmacokinetic profiles: a pilot study in children with congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2016) 2016:17. doi: 10.1186 / s13633-016-0035-5
PubMed Abstrakt | CrossRef Fulltekst/Google Scholar
47. Weintrob N, Dickerman Z, Sprecher E, Galatzer A, Pertzelan A. Ikke-klassisk 21-hydroksylasemangel i barndom og barndom: effekten av behandlingsstart på pubertet og slutthøyde. Eur J Endocrinol. (1997) 136:188–95. doi: 10.1530 / eie.0.1360188
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Bonfig W, Bechtold S, Schmidt H, Knorr D, Schwarz HP. Reduced final height outcome in congenital adrenal hyperplasia under prednisone treatment: deceleration of growth velocity during puberty. J Clin Endocrinol Metab. (2007) 92:1635–9. doi: 10.1210/jc.2006-2109
PubMed Abstract | CrossRef Full Text | Google Scholar
49. Dörr HG. Vekst hos pasienter med klassisk medfødt adrenal hyperplasi på grunn av 21-hydroksylase mangel. Horm Res. (2007) 68 (Suppl 5): 93-9. doi: 10.1159/000110587
PubMed Abstrakt | CrossRef Fulltekst/Google Scholar
50. Hoepffner W, Kaufhold A, Willgerodt H, Keller E. Pasienter med klassisk medfødt adrenal hyperplasi på grunn av 21-hydroksylasemangel kan oppnå målhøyde: Leipzig-opplevelsen. Horm Res. (2008) 70: 42-50. doi: 10.1159/000129677
PubMed Abstract | CrossRef Full Text | Google Scholar
51. van der Kaay D, van den Akker E. Ultralow-dose dexamethasone to preserve endogenous cortisol stress response in nonclassical congenital adrenal hyperplasia: a new promising treatment. Int J Endocrinol Metab. (2014) 12:e14657. doi: 10.5812/ijem.14657
CrossRef Full Text | Google Scholar
52. Lin-Su K, Vogiatzi MG, Marshall I, Harbison MD, Macapagal MC, Betensky B, et al. Treatment with growth hormone and luteinizing hormone releasing hormone analog improves final adult height in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. (2005) 90:3318–25. doi: 10.1210/jc.2004-2128
PubMed Abstract | CrossRef Full Text | Google Scholar
53. McCann-Crosby B, Chen M-J, Lyons SK, Lin Y, Axelrad M, Dietrich JE, et al. Nonclassical congenital adrenal hyperplasia: mål for behandling og overgang. Pediatr Endocrinol Rev. (2014) 12: 224-38.
PubMed Abstrakt/Google Scholar
54. Ogilvie CM, Crouch NS, Rumsby G, Creighton SM, Liao L-M, Conway GS. Medfødt adrenal hyperplasi hos voksne: en gjennomgang av medisinske, kirurgiske og psykologiske problemer. Clin Endocrinol. (2006) 64:2–11. doi: 10.1111/j.1365-2265. 2005. 02410.x
PubMed Abstract | CrossRef Full Text | Google Scholar
55. Mah PM, Jenkins RC, Rostami-Hodjegan A, Newell-Price J, Doane A, Ibbotson V, et al. Weight-related dosing, timing and monitoring hydrocortisone replacement therapy in patients with adrenal insufficiency. Clin Endocrinol. (2004) 61:367–75. doi: 10.1111/j.1365-2265.2004.02106.x
PubMed Abstract | CrossRef Full Text | Google Scholar
56. Diaz A, LAUFER MR Seteleie LLAA AV PC På EN, Vare AC Av O og GC PÅ AH. Menstruasjon hos jenter og ungdom: bruk menstruasjonssyklusen som et vitalt tegn. Pediatri. (2006) 118:2245–50. doi: 10.1542 / peds.2006-2481
CrossRef Full Tekst/Google Scholar
57. Merke DP, Poppas DP. Behandling av ungdom med medfødt adrenal hyperplasi. lancet Diabetes Endocrinol. (2013) 1:341–52. doi: 10.1016/S2213-8587(13)70138-4
PubMed Abstract | CrossRef Full Text | Google Scholar
58. Verma S, Green-Golan L, VanRyzin C, Drinkard B, Mehta SP, Weise M, et al. Adrenomedullary function in patients with nonclassic congenital adrenal hyperplasia. Horm Metab Res. (2010) 42:607–12. doi: 10.1055/s-0030-1253385
PubMed Abstract | CrossRef Full Text | Google Scholar
59. New MI. Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2006) 91:4205–14. doi: 10.1210/jc.2006-1645
PubMed Abstract | CrossRef Full Text | Google Scholar
60. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2018) 103:4043–88. doi: 10.1210/jc.2018-01865
PubMed Abstract | CrossRef Full Text | Google Scholar
61. Kruse B, Riepe FG, Krone N, Bosinski HAG, Kloehn S, Partsch CJ, et al. Congenital adrenal hyperplasia – how to improve the transition from adolescence to adult life. Exp Clin Endocrinol Diabetes. (2004) 112:343–55. doi: 10.1055/s-2004-821013
PubMed Abstract | CrossRef Full Text | Google Scholar
62. G. r., Bouvattier C, Duranteau L, Brauner R, Thibaud E, Kutten F, Et al. Nedsatt seksuell og reproduktiv utfall hos kvinner med klassiske former for medfødt adrenal hyperplasi. J Clin Endocrinol Metab. (2007) 92:1391–6. doi: 10.1210 / jc.2006-1757
PubMed Abstrakt | CrossRef Fulltekst/Google Scholar
63. Mercè fern Hryvndez-Balsells M, Muthusamy K, Smushkin G, Lampropulos JF, Elamin MB, Abu Elnour NO, Et al. Prenatal deksametason bruk for forebygging av virilisering i svangerskap med risiko for klassisk medfødt adrenal hyperplasi på grunn av MANGEL på 21-hydroksylase (CYP21A2): en systematisk oversikt og meta-analyser. Clin Endocrinol. (2010) 73:436–44. doi: 10.1111/j.1365-2265. 2010. 03826.x
CrossRef Full Tekst / Google Scholar
64. Bidet M, Bellanné-Chantelot C, Galand-Portier MB, Golmard JL, Sen V, Morel Y, et al. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2010) 95:1182–90. doi: 10.1210/jc.2009-1383
PubMed Abstract | CrossRef Full Text | Google Scholar
65. Birnbaum MD, Rose LI. Late onset adrenocortical hydroxylase deficiencies associated with menstrual dysfunction. Obstet Gynecol. (1984) 63:445–51.
PubMed Abstract | Google Scholar
66. Cuhaci N, Aydin C, Yesilyurt A, Pinarli FA, Ersoy R, Cakir B. Nonclassical congenital adrenal hyperplasia and pregnancy. Case Rep Endocrinol. (2015) 2015:296924. doi: 10.1155/2015/296924
PubMed Abstract | CrossRef Full Text | Google Scholar
67. Deneux C, Tardy V, Anne D, Mornet E, Billaud L, Charron D, et al. Fenotype – genotype korrelasjon hos 56 kvinner med ikke-klassisk medfødt adrenal hyperplasi på grunn av 21-hydroksylasemangel. J Clin Endocrinol Metab. (2001) 86:207–13. doi: 10.1210 / jcem.86.1.7131
PubMed Abstrakt | CrossRef Fulltekst/Google Scholar
68. Simonetti L, Bruque CD, Bregne Hryvndez CS, Benavides-Mori B, Delea M, Kolomenski JE, et al. CYP21A2 mutasjonsoppdatering: Omfattende analyse av databaser og publiserte genetiske varianter. Hum Mutat. (2018) 39:5–22. doi: 10.1002 / humu.23351
PubMed Abstract | CrossRef Full Text | Google Scholar
69. OMIM Entry – * 613815 – CYTOCHROME P450 FAMILY 21 SUBFAMILY A POLYPEPTIDE 2; CYP21A2.
Google Scholar
70. Miller WL, Witchel SF. Prenatal treatment of congenital adrenal hyperplasia: risks outweigh benefits. Am J Obstet Gynecol. (2013) 208:354–9. doi: 10.1016/j.ajog.2012.10.885
PubMed Abstract | CrossRef Full Text | Google Scholar
71. Lekarev O, New MI. Adrenal disease in pregnancy. Best Pract Res Clin Endocrinol Metab. (2011) 25:959–73. doi: 10.1016/j.beem.2011.08.004
PubMed Abstract | CrossRef Full Text | Google Scholar
72. New MI, Abraham M, Yuen T, Lekarev O. An update on prenatal diagnosis and treatment of congenital adrenal hyperplasia. Semin Reprod Med. (2012) 30:396–9. doi: 10.1055/s-0032-1324723
PubMed Abstract | CrossRef Full Text | Google Scholar
73. Kazmi D, Bailey J, Yau M, Abu-Amer W, Kumar A, Low M, et al. New developments in prenatal diagnosis of congenital adrenal hyperplasia. J Steroid Biochem Mol Biol. (2017) 165:121–3. doi: 10.1016/j.jsbmb.2016.06.016
PubMed Abstract | CrossRef Full Text | Google Scholar
74. Clayton PE, Miller WL, Oberfield SE, Ritzé EM, Sippell WG, Speiser PW, et al. Konsensuserklæring om 21-hydroksylasemangel fra European Society For Paediatric Endocrinology og lawson wilkins pediatric endocrine society. Horm Res. (2002) 58:188-95. doi: 10.1159 / 000065490
PubMed Abstrakt | CrossRef Fulltekst / Google Scholar
75. Nandagopal R, Sinaii N, Avila NA, Van Ryzin C, Chen W, Finkielstain GP, et al. Fenotypisk profilering av foreldre med kryptisk ikke-klassisk medfødt adrenal hyperplasi: funn i 145 ikke-relaterte familier. Eur J Endocrinol. (2011) 164:977–84. doi: 10.1530/EIE-11-0019
PubMed Abstrakt | CrossRef Fulltekst / Google Scholar
76. Takasu N, Nakachi K, Higa H. Utvikling Av Graves ‘ hypertyreose forårsaket en binyrekrise hos en pasient med tidligere ukjent ikke-klassisk 21-hydroksylasemangel. Intern Med. (2010) 49:1395–400. doi: 10.2169 / internalmedisin.49.3573
PubMed Abstract | CrossRef Full Text | Google Scholar
77. Meyer-Bahlburg HFL, Dolezal C, Baker SW, Carlson AD, Obeid JS, New MI. Prenatal androgenization affects gender-related behavior but not gender identity in 5-12-year-old girls with congenital adrenal hyperplasia. Arch Sex Behav. (2004) 33:97–104. doi: 10.1023/B:ASEB.0000014324.25718.51
CrossRef Full Text | Google Scholar
78. Meyer-Bahlburg HFL, Dolezal C, Baker SW, NEW MI. Seksuell orientering hos kvinner med klassisk eller ikke-klassisk medfødt adrenal hyperplasi som en funksjon av graden av prenatal androgenoverskudd. Arch Sex Oppfører Seg. (2008) 37:85–99. doi: 10.1007 / s10508-007-9265-1
PubMed Abstract | CrossRef Full Text/Google Scholar