定義と有病率
先天性副腎過形成(CAH)は、コルチゾール産生に必要な5つの副腎ステロイド生成酵素のいずれかの欠乏による軽度から急性コルチゾール合成障害を特徴とする常染色体劣性疾患のファミリーを包含する(1、2)。 従来、CAHは、(a)塩消耗または出生時および/または新生児期に現れる単純なウイルス化形態を呈する古典的(CCAH)と、(b)非古典的(NCCAH)に分けられ、生殖器のあいまいさを欠いていて、すぐに生命を脅かすことはなく、後の人生で提示される、または無症候性のままである、または別の疾患として誤診される(3)。 しかし、CAHの異なる形態にわたる境界は厳密には定義されておらず、これはこの障害に関連する課題を増加させる傾向がある。 したがって、重度から軽度または他の無症候性(に、表現型の連続体としてCAHを考慮することをお勧めします3)。
CAHの最も一般的な形態は、21-ヒドロキシラーゼ欠乏症(21OHD)によって引き起こされ、17ヒドロキシプロゲステロン(17OHP)が11デオキシコルチゾールに、プロゲステロンがデオキシコルチコステロンに変換されないか、または変換されない。 ステロイドの転換の封鎖は男性ホルモンの前駆物質の高められた生産で、CRH-ACTHの刺激の下で起因し、生化学的なhyperandrogenismをもたらします、上昇した17-OHPのレベ この病気の古典的な形態は、世界中の16,000人の生児のうち1人に発生します。 しかし、疾患の知覚される発生率は、使用されるスクリーニング方法に関連していることに言及すべきである。 例えば、スウェーデンのCAHの古典的な形態の発生率はケースの調査のアプローチが使用されるとき1:11,500であることが報告されました、しかし図は1:9,800 同様に、フランスとスイスでの発生率は、スクリーニングの異なる方法が使用されている場合、1:15,472と1:23,000と1:10,749と1:11,661の間の範囲である(4)。
NCCAHははるかに頻繁に発生し、約1,000人の白人のうち、アシュケナージ-ユダヤ人(1:27)、ヒスパニック(1:53)、ユーゴスラビア(1:62)、イタリア人などの特定の民族グループでより一般的に発生している。(1:300) (5, 6). それにもかかわらず、異なる推定方法(症例調査、ホルモンスクリーニング、および分子検査)に基づくNCCAHの有病率に関するデータは欠けている。 NCCAHは、1:25から1:10のキャリア頻度を有する最も一般的な常染色体劣性内分泌障害と考えられている。 21-OHDによるNCCAHでは、残留酵素活性は約10-70%(7-9)であると推定される。
診断
生殖器のあいまいさおよび/または塩消耗症状のために出生時または新生児期に行われる疾患の古典的な形態の診断と比較して、またはNCCAHのほとんどの症例が容易に検出できない国もある(4、10)。 さらに、多くの個体は、小児期および青年期に無症候性のままであり、正常な生殖機能を有し、他の家族の診断および結果としての検査(11)のためにNCCAHに 但し、NCCAHのほとんどの女性は男性ホルモンの余分な物の徴候を経験し、臨床疑いがテストを促すとき、高い基礎17OHPのレベルがNCCAHの診断を指しないよ
確かに、内分泌学会によって提案された臨床ガイドラインは、nccahのスクリーニングツールとして17OHPのベースライン非刺激値を推奨しています。 朝17OHPレベル>6nmol/L以上のレベルがNCCAH個人の90%をキャプチャすることが示されているので、月経中の女性の濾胞期における6nmol/L(12、13)。 これらは、多くの場合、NCCAH患者では正常なレベルをもたらすので、17OHPのランダムな測定は、有用であることが示されていない、と彼らは、さらに、健康な女性(の半分の黄体期に非常に高いです13)。 しかし、この疾患を有する280人の被験者から得られた我々のデータでは、6人の患者(2。1%)は、ベースライン17OHP値<6nmol/L(14)を有していた。 さらに、Bidet e t a l.、分子技術によって検証されたNCCAHを有する女性の大規模なコホートでも、研究対象の8%において6nmol/Lよりも低い基礎17OHP値を発見した(15)。 最後に、Speiserらによって収集されたデータに基づく。、NCCAHを有する個体の9%は、2ng/ml(6nmol/Lに相当する)よりも低い17OHP値を示した。 他の研究によると、5.1と9nmol/Lの間の17OHPのベースライン値は、NCCAH(の診断のために十分である13、16、17)。 最近では、基礎のレベル17のOHPの4。6nmol/Lは、小児期(18)の間に早期adrenarcheと被験者におけるNCCAHを予測するACTHテストのためのしきい値として示唆されました。
これらの知見と提案の合計は、Synacthen刺激試験を受ける患者の選択がケースバイケースで評価されるべきであることを示している。 診断には3nmol/Lを超える17OHPの刺激後レベルが必要です(19)。 影響を受けていない被験者で重複があるものの、一つのCYP21A2変異を運ぶヘテロ接合体は、ACTH刺激後わずかに上昇した17OHPレベルを示す(9)。 しかし、Dacou-Voutetakis et al. 基礎および刺激後の17OHP値の合計が1.5nmol/Lを超える場合、ヘテロ接合性の可能性は非常に高いことを指摘している(20)。
もう一つの重要な考慮事項は、17OHP評価に使用される技術に関するものです。 17OHPレベルは、様々な免疫測定法によって測定されるが、最近示されているように、最も正確で信頼性の高い結果は、液体クロマトグラフィーと質量分析(LC-MS/MS) 実際、LC-MS/Ms測定を標準的な方法(21)と比較したときに多くの偽陽性17OHP測定が発見されたにもかかわらず、後者の手順は普遍的に使用されておらず、不確実な診断が発生した場合には、より正確な結果が得られる。 注目すべきことに、定量化されたLC−MS/MS1 7OHPのスクリーニングおよび診断閾値は、まだ定義されていない。 これらの閾値は、LC−MS/MSの特異性の増強、交差反応性および干渉を受けにくい方法(2 2、2 3)のために、イムノアッセイで確立されたものよりも低い可能性が 確定診断に尿中ステロイドプロファイルが必要かどうかは解明されていない(22)。 境界例では、他の酵素の欠陥からの21ヒドロキシラーゼの不足を区別し、しっかりした診断を確立するためにACTHの刺激テストの後で完全なadrenocorticalプロフ
具体的には、21-ヒドロキシラーゼ欠乏症を確認し、他の酵素検出を排除するために、完全なステロイドプロファイルを曖昧な場合に行うべきである。 血清ステロイドプロファイルに17OHP、コルチゾール、11-デオキシコルチコステロン、11-デオキシコルチゾール、17-OH-プレグネノロン、デヒドロエピアンドロステロン、アンドロステンジオンを含めることは、11β-ヒドロキシラーゼ欠乏症、よりまれに3β-ヒドロキシステロイドデヒドロゲナーゼ欠乏症またはP450オキシドレダクターゼ欠乏症(24)などのCAHの他の原因を排除するのに有用である。 遺伝子検査は、現時点ではNCCAHのための主要な診断ツールであるとは考えられていないが、それは診断の確認と遺伝カウンセリングのために必須である(3)。
コルチゾールレベルに関する限り、一般に少なくとも496nmol/Lの刺激後の値が期待される。
コルチゾールレベルに関する限り、一般に少なくとも496nmol/Lの刺激後の値が期待される。
注目すべきは、Stoupa e t a l.、60%の21OHDの結果としてNCCAHを持つ47人の子供は、刺激後に低いコルチゾール値を持っていた、主要なストレッサー事象(中に副腎不全の開発のための増加したサーベイランスの必要性を指している発見25)。女性におけるNCCAH発現NCCAHの臨床発現は、発症年齢だけでなく、異なる徴候および症状にも関係するため、高レベルの多型を特徴とする。
NCCAHの臨床発現は、発症年齢だけでなく、異なる徴候および症状にも関係している。 NCCAHの最初の臨床提示は、10歳前の症例の11%、10歳から40歳までの症例の80%にあることが報告されています(12)。 CAHとNCCAHにおける遺伝子型-表現型相関はまだ解明されていない。 Speiser et al. CYP21A2(26)の対立遺伝子の変化から21-ヒドロキシラーゼ欠乏症の結果の表現型の変動のほとんどが、すべてではないことを示唆しています。
小児期の症状
新生児期には、典型的にはNCCAH女性は無症候性のままであり、正常な外部生殖器を有する。 報告されたNCCAHの最も初期のケースは、陰毛を開発した6ヶ月の少女(11)です。 軽度のコルチゾール欠乏症はまた、いくつかのケースで発生する可能性がありますが、通常、NCCAH症例における臨床所見および症状は、5歳以降、さらには、増加したアンドロゲンレベルに関連しています(8)。 NCCAH症例の92%において、最初に現れる症状は時期尚早pubarche(PP)であり、これは女児では8歳未満、男児では9歳未満(12)であり、推定有病率は10〜11.3%(15)である。 注目すべきは、PPと呼ばれる小児では、異なる研究によると、NCCAHの発生率は5〜30%の範囲である(20、27)。 しかし、NCCAHの分子確認を有する280人の患者の分析に基づいて、94歳未満の女性におけるPPの発生率は88%と同じくらいであることがわかった。 一方、NCCAHを有する45人の男性の分析では、被験者のわずか29%でPPが同定された(14)。 さらに、我々はPPを持つ被験者の約半分がCYP21A2変異のヘテロ接合体キャリアである可能性があることに留意すべきである。明確化が必要なもう1つの側面は、PPと早熟思春期との関係です。
場合によっては、エストロゲンへの副腎の男性ホルモンの周辺芳香族化がhypothalamic下垂体生殖腺の軸線を活動化させるかもしれないことが二次早熟な思春 そのような場合には、定期的なフォローアップが必要である。 何人かの子供は多毛症、アクネ、および/または急速な成長を開発し、高い構造および骨の年齢の進歩を明示します(29)。 後者は、急速な骨端融合の結果として切り捨てられた最終的な高さをもたらす可能性がある。 小児期の他のまれな臨床所見には、唇の癒着、肛門周囲の毛、陰核肥大、および声の嗄声(30-32)が含まれる。思春期および成人期における症状
思春期および成人期には、女性のNCCAHは多嚢胞性卵巣症候群(PCOS)に似た高アンドロゲン症状を示す(33)。 症状には、多毛症、座瘡、アンドロゲン性脱毛症、無排卵、月経機能不全、および不妊症が含まれる。 多施設研究では、思春期および成人女性の間で最も一般的な症状は、多毛症(59%)、乏月経(54%)、および座瘡(33%)であった。 NCCAHを持つ161人の女性のうち、症状を提示することは、多毛症(78%)、月経機能障害(54.7%)、および不妊(12%)の減少(34)であった。 疾患を有する161人の女性の私たちのグループでは、患者の63%が多嚢胞性卵巣様表現型(14)を提示した。 高アンドロゲン血症の女性ではNCCAHの発生率が高いため、この所見は逆の意味を持つ。 確かに、Witcelらによる研究。 高アンドロゲン性女性における1〜33%の範囲のNCCAHの発生率を報告することは大きな関心事である。 アンドロゲン過剰量の評価のために紹介された950人の女性の別の研究では、NCCAHはそれらの4.2%で検出された。 しかし,NCCAH患者は他の高アンドロゲン患者群と比較して若く,より多くのhirsuteであった。 それらはまた他のhyperandrogenicシンドロームの患者よりテストステロン、自由なテストステロンおよび17OHPのかなりハイレベルによって特徴付けられました。 卵巣超音波では、それらの77%が多嚢胞性卵巣を示し、41%が卵巣の大きさを増加させた。 要約すると、NIHとRotterdamによるPCOS基準をそれぞれ56と72.8%の割合で満たしていました(35)。
この疾患の進行性は、多毛症の有病率が年齢とともに増加することが示されており、思春期前にはまれであることが観察されているという事実に 多毛症は、NCCAH患者で報告されている最も一般的な臨床症状であり、71〜96%(28、36)の範囲である。 NCCAHを持つ思春期の女の子は、典型的には多毛症(と存在37、38)。 スペクトルのもう一方の端には脱毛症の問題があります。 NCCAH患者では男性型脱毛症が報告されている。 脱毛症の有病率はまた、年齢とともに増加するように見えました,彼らの人生の第二十年の間に患者の6%から第五の19%に,再び病気の進行性の性質を示 にきびはNCCAH被験者(のほぼ33%で報告されている36)。 驚くべきことに、NCCAHを引き起こすCYP21A2遺伝子の変異は、重度のにきび(40)を提示する4.9%の123成人女性で検出されました。
oligomenorrheaまたは続発性無月経を含む月経不順は、多くの場合、初潮後の個人におけるNCCAHの提示徴候であり得る。 さらに、症例の8-9%も原発性無月経を経験し、この理由から、初めて医師の診察を受ける。 さらに、NCCAHの個人の間で、oligomenorrheaは第一次無月経より共通です。 例えば、Moran e t a l.、10歳から19歳のNCCAH女性の間で、oligomenorrheaは、原発性無月経(12)と思春期のわずか9%と比較して、ケースの56%に存在していました。 結論として、非常に多くのNCCAH症状はPCOSの症状に似ているため、NCCAHがPCOSの大きなミミッカーと呼ばれていることは驚くべきことではありません。 いずれにしても、高アンドロゲン症状のために紹介された若い女性の評価中にNCCAHの診断を考慮する必要があります。Arltらによる研究では。
、NCCAH患者は、一致したコントロール(と比較して有意に高いBMIと低い最終的な高さによって特徴付けられた41)。 これらのデータは、インスリン非感受性の存在を示すいくつかの研究と併せて、アンドロゲン濃度の上昇の効果および/またはグルココルチコイド療法の結果として病態生理学的に説明され得る好ましくない代謝プロファイルを示唆している(42,43)。 骨粗鬆症は、おそらくコルチコステロイド療法の結果として、これらの被験者においても検出することができる(34)。
最初の症状から成人生活までのNCCAH管理
NCCAHの診断が確立されると、その後の治療勧告に関連するいくつかの問題が考慮されます。 対処すべき最初の質問は、グルココルチコイド(GC)療法が示されているかどうかである。 このアプローチの後ろの一般的な推論は患者に十分なコルチゾールの集中の提供によって、彼女の毎日の必要性がカバーされ、従って、CRH ACTHの軸線の刺激が先を細くされ、減らされた副腎の男性ホルモンの生産をもたらすことである。 このアプローチに関する一般的なルールは、年齢、性別、実験室データ、患者固有の推奨事項、およびグルココルチコイド補充療法に対する治療の目標の影響が真剣に考慮されているが、GCsは薬理学的用量ではなく置換時に与えられるということである。 しかし,GC置換療法の長期化は高コルチゾール血症につながり,代謝のあらゆる側面,特に成長,脂肪分布,インスリン抵抗性,ならびに心理的プロファイルに対する十分に文書化された悪影響をもたらす可能性があることにも留意すべきである。 このアプローチの別の主要な不利な点は甲状腺機能低下症のTSHの価値に関する存在のような十分な取り替えの適量の十分な臨床索引か生化学的なマー
最後に、GCを使用すべきコンセンサスがなく、GC投与の異なるモダリティに関する長期的なデータがないことは、主治医の決定と各患者の最適な選択の選択をさらに複雑にするという事実である。 Hydrocortisoneは子供で最も密接に自然なホルモン(コルチゾール)に類似しているが、多数の毎日の投薬のための必要性による青年および若い女性の適したアプロー それ故に、ほとんどの大人のendocrinologistsは適切な線量でdexamethasoneかprednisoloneを、好みます。 一方、異なるGcの等価性は、それらの抗炎症作用に基づいており、ヒト代謝の異なる側面に基づいていないことを指摘しなければならない。 コルチゾールはヒトゲノムのほぼ20%に影響を及ぼすため、様々な組織において異なるGcの多様な応答が期待される。 この認識を裏付けることは、CAH患者へのデキサメタゾンの投与が他のGCsと比較してインスリン抵抗性の指標の低下を示しているという事実である(44)。 さらに、プレドニゾロンの2.5–7.5mgの管理、常態として考慮される線量は骨の新陳代謝(の長年の否定的な影響を出します45)。 従って、ヒドロコルチゾンはGCの補足療法の場合の処置の最もよい形態考慮されるべきです。 新しく合成されたヒドロコルチゾン式の出現と、CAH患者における最初の肯定的な結果は、将来のために多くの約束を示しています(46)。
小児期の管理
小児期にPPの徴候を有すると診断されたNCCAH患者は、副腎ホルモンを抑制し、最終的な身長に影響を与える可能性のある骨年齢の急速な進行を防止することを目的として、ヒドロコルチゾンで治療することができる。 ヒドロコルチゾン治療が思春期の発症の1年前に開始され、9歳以下の骨年齢を有する患者では、最終的な高さは遺伝的可能性の範囲内にとどまった(47)。 利用可能な研究は、成人の身長が密接に監視され、投薬計画(48-50)に厳密に準拠している患者の予想される目標身長に近づいたことを示しています。 5つのケース(患者6.1–9.2年齢)の最近の小さい調査はデキサメタゾンの超低い線量が内生圧力および効果を減らす有望な選択であることを示しました。 これが現実的なアプローチを構成するかどうか、および投与される用量がどのくらいであるべきかは解明されていない(51)。 Nebesioらによる研究。 CCAHの9つのprepubertal患者のヒドロコルチゾン、prednisoloneおよびdexamethasoneのホルモン性の効果そしてpharmacokineticsを比較しました。 彼らはdexamethasoneがdexamethasoneが副腎の男性ホルモンの生産(46)の抑制のためにより有効であることをかなりより低い副腎のホルモンのレベルの達成の他の形態、そし もちろん、この発見を検証または拒否するにはさらなる研究が必要です。 なお、場合によっては、高い男性ホルモンの集中は早期の思春期の原因となるGnRHの軸線の二次刺激をもたらすかもしれません。 このような場合、骨年齢が年代年齢よりも有意に高い場合、および/または予測された最終身長が目標身長に不釣り合いである場合、GnRHアナログとの平行
もう一つの重要な考慮事項は、GnRH類似体およびGH処方による治療が目標高さ(52)の達成をもたらした場合に付随するGH欠乏症の症例に関する。 現在のガイドラインによれば、上記の治療は、予測された高さSDが-2.25<目標の高さまたはそれよりも低い(53)場合にのみ慎重に推奨される。 ヒドロコルチゾン治療を開始するための代替適応症は、ACTH刺激後の不十分なコルチゾール応答である(36)。
小児における好ましいGC治療は、通常、ヒドロコルチゾン10-15mg/m2であり、三つの用量に分けられる。 多くの場合、低用量はまた、6mg/m2/日(から開始して、有効であることが証明されている34)。 過剰摂取はcushingoidの特徴と同様、悪い成長で起因できるそれを考慮する避けるべきです。 規則的な成長パターン、年代順の年齢に互換性がある骨の年齢、および中央肥満の不在はまた適切な管理のための臨床索引として役立つかもしれません。 生化学的な/ホルモン性のプロフィールに関して、骨の年齢のための上部の範囲への中間のandrostenedioneおよびテストステロンのレベルはNCCAHの子供の十分なGCの しかし、抑制されたテストステロンレベルはNCCAH患者の10%で発見されたが、患者の別の28%がテストステロン濃度を増加させていたのに対し、この現象は DHEASの価値は17OHPおよびプロゲステロンのレベルの抑制が非常に高いGCの線量を要求する一方、非常に小さい線量と減ります。 17のOHPの価値が診断のために重大であるが、フォローアップの間に有用ではないことに注意されるべきです。 注目すべきは、ホルモン評価のための採血は、治療を中止することなく実施されなければならないことである。 しかし、病気の複雑さとその多面的な性質のために、小児におけるレジメンの変更またはグルココルチコイド療法の中止のタイミングに関する特
思春期の管理
小児期以降に治療された患者
NCCAH女児における正常な月経パターンが確立されるまで、小児期に開始されたGCsの継続 しかし、青年期の患者は、しばしば薬物の慢性投与に十分な遵守を示さず、しばしば用量を省略する。 さらに、思春期の間に、ヒドロコルチゾンの半減期は50%によって11º ohsdの活動を減少する、また増加されたコルチゾールの整理が原因で増加されたIGF-1レベ 若い女性では、2年のポストの初潮および正常な排卵周期が記録されれば、現われるかもしれないhyperandrogenic徴候の方の患者中心のアプローチは強く推薦され
多毛症、座瘡、および/または乏月経などの重度の高アンドロゲン性所見がある場合は、治療の継続が考慮されます。 若い思春期の脆弱な感性を覚えておくことは重要ですが、これは高アンドロゲン症の”忌避”徴候によって深刻な損傷を受けるでしょう。 Hyperandrogenic印、menstrual無秩序および生活環境基準の悪い組合せはよく文書化され、より若い女性で特に高いです。 考慮すべきもう一つの点は、ACTH刺激後の副腎予備の応答である。 刺激後の思春期および成人女性のピークコルチゾールが496nmol/L以下であることを考えると、ストレスの場合には、ステロイド治療を投与すべきである(58)。 一方では、上記の徴候のどれも若い患者で見つけられなければ、GCの処置は規則的なフォローアップが助言される後で中断されるかもしれません。
思春期の最初の症状または治療中止後の高アンドロゲン症状を有する女性
高アンドロゲン症状の存在下では、患者の主な愁訴に焦点を当て、患者指向のアプローチが強く推奨される。 これは、後者のアプローチでは、若い患者が失望し、これが治療の中止につながり、その結果、成人期の有害な健康転帰につながるので、一般的な普遍的な 重度の多毛症の場合、経口避妊薬(Ocp)を用いた6-12ヶ月のコースは、OcpがSHBG肝臓産生および卵巣からのアンドロゲン放出の減少に有益な効果を与えると、合理的な手順を構成する。 臨床改善は最低のandrogenic特性が付いているprogestagenの同時使用は強く推奨されているが、OCPsの開始の少なくとも2-3か月に続いて期待されます。 OCPsが最初のラインアプローチとして失敗すれば、antiandrogens(spironolactone、flutamide、bicalutamide、cyproteroneのアセテートおよびfinasteride)は処置に加えられるかもしれません。 我々の経験では、ビカルタミドの投与は重度の座瘡の症例で有意な改善を達成したが、重度の多毛症の症例では同様の結果は得られなかった。 このようなレーザーアプリケーションや脱毛剤などの化粧品のアプローチはまた、過剰または不要な髪の成長や頭皮の脱毛(アンドロゲン性脱毛症)を訴える女性のために提案することができます(32、34、57)。刺激後のコルチゾール分泌が不十分な患者、またはOCPsおよび抗アンドロゲンが許容できない場合は、GCsによるコースが強く推奨されます(57)。 New e t a l. 多毛症は、ほぼ30ヶ月(必要とするのに対し、不規則な月経とにきびは、グルココルチコイド療法の開始後3ヶ月以内に逆転していることを示しています59)。 小児期とは対照的に、青年期には、長時間作用性ステロイドがしばしば使用され、プレドニゾロン5mgまたはデキサメタゾン0.25mgのレジメンが推奨さ しかし、現実の世界では、臨床データはNCCAH管理に適用される様々な異なるレジメンを示している(41)。
治療の主な目標は、高アンドロゲン症状からの患者の救済でなければならない。 したがって、検査結果のみに基づいて診断されたNCCAHを有する個人の間では、ホルモンおよび分子所見は必ずしも臨床的重症度を予測するわけではな 内分泌学会ガイドラインによると、NCCAH患者は、症状が解決したときにGC療法を中止する選択肢を与えられるべきである(60)。 もちろん、これらの患者はフォローアップに失われるべきではなく、症状の再発の場合には治療を再開始すべきである。 さらに、中止の場合、患者は不妊症の可能性について知らされるべきであり、妊娠したい場合は医師の診察を受けることを奨励されるべきである(19)。 注目すべきことに、小児から成人内分泌専門医への患者の適切な移転は、1年間の同期モニタリング(53、61)の後に最適に実施されるべきである。
NCCAHと生殖
亜受精
亜受精は、主に月経障害、慢性無排卵、および解剖学的変形による古典的な形態のCAHで一般的に報告されている。 出生率は対照人口(の65%と比較して17%と推定されている62)。 一方、NCCAH女性の不妊に関するデータは最近詳細に評価されており、推定不妊症発生率はNCCAH女性の11%であり、CAHよりも比較的軽度であり、多くの場合容易に解決されている。 Bidet et al. NCCAHに罹患している190人の女性の不妊治療を評価し、そのうち95人が妊娠したいと考えていました。 この集団では、85人の女性で187回の妊娠が報告され、82人で141回の出産が報告されました。 NCCAHの診断前に99の妊娠(52.9%)が発生し、そのうち3つは排卵誘導プロトコルを適用し、残りは自発的であり、88の妊娠はNCCAH診断後に行われたことを強調しなけ それらの大部分では、妊娠はヒドロコルチゾンによる治療の施行後に発症したが、11人の女性では自発的に起こった(63、64)。 妊娠を望む22観察NCCAHの女性の別の報告では、12の妊娠はプレドニゾン(65)で続いた。
流産
Bidet et al.のコホートにおける流産
Bidet et al.のコホートにおける流産
流産率は、gcsで治療された患者および未治療の患者でそれぞれ6.5および26.3%であった(64)。 Cuhaciらによる症例報告でも報告されているように、NCCAHがまだ診断されていない場合、妊娠の結果はグルココルチコイド治療なしで成功する可能性があ それにもかかわらず、同じカップルは、2つの流産を経験し、この最初の妊娠(66)後に亜受精を報告しました。 早産妊娠の可能性に関する限り、Bidet et al.、それは人口(の残りの部分のそれと大きく異ならない15)。
不妊計画
症候性高アンドロゲン症または報告された不妊症を有するが、妊娠したい女性のために、GC療法を強くお勧めします。 この期間中、使用されるグルココルチコイドはプレドニゾンまたはヒドロコルチゾンである。 それはその後のステップを導くので、最も重要なアクションは、将来の両親の遺伝子検査です。 妊娠することを望むNCCAHの女性は病気の古典的な形態の幼児を出産する危険に気づいているべきです。 NCCAHを持つ女性の間で妊娠の結果、より具体的にはCCAHで生まれた乳児の発生率は、約15%(NCCAHで、1.5と2.5%の間であることが最近の研究で推定される36、64)。 もちろん、この頻度は、参照集団、高い結婚率(と集団で発生するより高い発生率に依存する36)。 CAHを持つ子供を出産する親の予測可能性は、未知の父方の遺伝子型の場合は120に1であり、父親に関連する変異がない場合はゼロであり、ヘテロ接合性変異がある場合は4に1である(19)。 その結果、これらの女性のパートナーの遺伝子検査は、CAH(の古典的な形で子供を出産するリスクを評価するために不可欠である64、67)。
将来の母親が2つのNC変異を有する場合(表1)、CYP12A2変異のヘテロ接合体である場合、子孫は将来非古典的な形態の疾患を発症する危険性があ しかし、キャリアの状態は異なる集団で2-15%であり、これらの個体の半分は重度の突然変異を有することに留意すべきである。 さらに、遺伝子検査が必要とされない状況に関する決定的な勧告は、特に軽度の変異のために、不完全な遺伝子型-表現型相関を考えると困難である。 例えば、NCCAHに最も頻繁に関連するP30L変異は、古典的なCAHを有する患者においても生じる。 大規模な多拠点ヨーロッパの研究は、最近、これが患者の78%の場合であることを示した。 本研究では、軽度のV218L変異は、症例の30%においてNCCAHではなく古典的なものと関連していた。 したがって、NCCAHを有するすべての患者は、生殖パートナーの遺伝カウンセリングおよび分子評価を提供すべきである。テーブル1
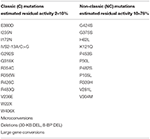
最小(C)または中程度(N C)の残留酵素活性を有するCYP2 1A2の変異(1 4、6 8、6 9)。対照的に、将来の母親が1つの重度の(C)および1つのNC変異を有する複合ヘテロ接合体である場合、将来の父親の遺伝子検査は必須である。 私たちは、イムノアッセイまたはより正確なLC-MS/MS手順を使用して研究から発信されたデータは、一貫して確実に基底またはACTH刺激17OHP値を使用してヘテ Synacthen試験の使用を示唆する可能性があり、それがヘテロ接合性(基底およびピーク刺激17OHP値の合計<1.5nmol/L)と互換性がない場合、DNA検査は避けられ しかし、他の研究では検証されていないため、このテストの解釈には非常に慎重でなければなりません。 したがって、ACTHの使用は、単独またはステロイド比またはステロイドプロファイルの一部として21-デオキシコルチゾールを刺激し、特にLC-MS/MSがより広 将来の父親がNC変異を持っている場合、他には何も必要ありません。 逆に、彼が古典的な突然変異のキャリアである場合、胎児の出生前治療に関する問題が生じる。 すべての可能な遺伝子型の組み合わせおよび提案された手順を表2に示す。テーブル2

将来の両親の遺伝子型に基づいて、妊娠中の提案された手順の計画。
CAHの出生前治療
CAHの出生前治療の目的は、胎児の生殖器の男性化の予防だけでなく、あいまいな生殖器(70)を持つ子供を持っている可能性があ Dexamethasoneは胎盤を交差させる機能のためにそして胎盤がある11β OHのステロイドの脱水素酵素から非活動化させないし、母の血のコルチゾール結合のグロブリン(CBG)にだけ最小に結合するので使用されます。 デキサメタゾンは、胎児のACTHの分泌の抑制によって、高められた胎児の男性ホルモンの生産を減らします。 核型またはDNA分析が男性または影響を受けていない女性をそれぞれ明らかにした場合には、治療を中止しなければならない(71)。 それにもかかわらず、胎児の生殖のvirilizationが6−7週のポストの概念で既に始まったが、遺伝子診断のための絨毛性絨毛のバイオプシーは10-12週の前に得 この時間間隔は、8胎児のうち1だけが影響を受け、女性であっても、CAHをウイルス化する危険性のあるすべての妊娠が治療されるべきであることを示唆しています(19、34、70)。
予防措置のための胎児のデキサメタゾンへのこの曝露が医学的および倫理的に許容されるかどうかは、議論の余地がある(70)。 動物実験では、妊娠初期のデキサメタゾンが出生体重を減少させ、腎膵臓ベータ細胞の機能と脳の発達に影響を与え、成人期の不安、高血圧、高血糖の素因となることが実証されています(70)。 なお、妊娠の間のdexamethasoneの処置を使用して妊婦の10-20%は体重増加、高められた食欲、気分のむら、不眠症、浮腫、高血圧、hyperglycemiaおよび脈理に不平を言います。 しかし、New e t a l. 出生前にデキサメタゾンで治療された胎児における平均Praderスコアよりも低いが、長期的な結果に差はないことが判明した(72)。 さらに、Merce Fernandez-Balsells et al.、デキサメタゾンは重要な母性的なか胎児の悪影響なしで胎児のvirilizationの減少と関連付けられるために示されていました(63)。 しかし、上記のレビューの著者だけでなく、内分泌学会の連続ガイドラインは、文献の全身の小さなサンプルサイズのために、主題は不確実なままであり、さらなる調査が明らかに必要であることを指摘している。
治療を開始することについての決定は、経験豊富なチームと制度的審査委員会によって承認され、家族の価値観や好みに基づいて、前提条件(19、63)とし この分野のいくつかの専門家は、両親や患者にあいまいな生殖器の感情的な通行料を課すのではなく、母親と胎児のデキサメタゾンへの不必要な出生前の暴露を防ぐことに高い価値を置くべきであることを示している。 最も重要なことは、デキサメタゾン投与は患者の状態を変更するのではなく、生命や知的能力を維持するのではなく、手術の必要性を減らすことに向 彼らはまた、彼らの子供が治療にもかかわらず、病気の古典的な形態に苦しむ可能性が高いことを知っている必要があります。 一方、最も重要なことに、胎児のプログラミングと成長の特に敏感な期間中に非常に強力なGCへの若い生物の暴露は、XY核型を持つ胎児の場合には役に立たないことを証明するかもしれませんが、現在のところ、堅牢で疑う余地のないデータによって支持されていません。
理想的には、早期診断が必要であり、これは非侵襲的な技術を介して、胎児の生殖器の男性化の開始前に行われるべきである。 この方向で働いて、新しい研究は、胎児の性別の決定と早期妊娠期(<9週間)でのCAHの診断を可能にする有望な方法として、母体血漿から得られた無細胞胎児DNAの使用を指摘している。 この手順が臨床現場で広く実施されている場合、不必要な出生前デキサメタゾン治療は回避される(73)。
妊娠中の治療
妊娠中には、NCCAHの女性は、通常、妊娠がストレスレベルの上昇を伴うだけでなく、上記の流産発生率の高い予防措置として、ヒドロコルチゾンで治療することが好ましい。 この形態のグルココルチコイドは、胎盤酵素11β OHステロイドデヒドロゲナーゼIIによる代謝のために支持され、グルココルチコイドが胎児に影響を及ぼすことを防止する(71)。 概念の時に、ヒドロコルチゾンの線量は20-25mg/dayに高められるべきで、線量の修正は妊娠の学期(74)の上部の正常なレベルでテストステロンのレベルを保 一例として、2人の健康な双子の出産後3年までの受胎からのNCCAH患者のテストステロンおよびD4値が示されている(図1)。
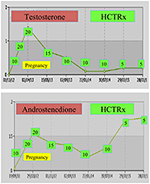
図1. Nccahを持つ患者に管理される3年のポスト配達までの概念からのテストステロンおよびandostenedioneのレベルそしてそれに続くヒドロコルチゾンの投薬(HCTRx)。
その他の特別な問題
NCCAHにおけるストレス管理
主要な生命を脅かすストレス、手術、または重篤な病気の間に、グルココルチコイド治療されているNCCAH患者は、副腎機能が医原性に抑制されていることを考えると、グルココルチコイドのより多くのまたはより頻繁な用量を必要とすることがある。 従って幼児の親を教育することは重大、また圧力の投薬についての大人の心配への彼らの転移の患者を、再教育するためにである。 GCsで治療されていないNCCAH患者または中止の場合、ACTHに対するコルチゾール応答を評価する必要があります。 NCCAH患者のほぼ3分の1は、刺激(に不十分に応答する15、75)。 ACTH刺激に正常に反応する人には、ストレス用量での治療は推奨されません(19)。 これらの患者は正常なアルドステロン分泌を有し、塩消耗を発症しないので、ミネラロコルチコイド療法はいずれの場合も必要とされない(58)。 注目すべきは、甲状腺機能亢進症またはレボチロキシン治療の増加の場合、コルチゾールクリアランスが増加し、副腎の危機が起こることがある(58、76)。
NCCAHにおける心理生物学的側面
Meyer-Bahlburg et al. いくつかの研究では、21OH欠乏症によるNCCAH患者の心理的プロファイルの障害が報告されています。 より具体的には、影響を受けた女性は、正常な対照女性と比較して、ジェンダー関連行動のいくつかの尺度で高い男性化/defeminizationスコアを有していたが、古典的なCAH 彼らはまた、バイセクシュアルまたは同性愛者の指向を有意に増加させた。 これらの女性とのレトロスペクティブ臨床定性的インタビューは、にきび、多毛症、および受胎困難(などのアンドロゲン依存性の兆候、との彼らの前処 同様に、Arlt e t a l. 主観的健康状態が有意にドメインに関連して、年齢と性別にマッチしたコントロールと比較して、最も顕著な違いで、短期的な健康調査のすべての 病院の不安とうつ病スケール(HADS)アンケートはまた、増加した不安スコア(41)を明らかにした。 これらの知見を念頭に置いて、治療を導くための心理的パラメータは、NCCAHの女性で考慮されるべきであり、患者指向のアプローチの文脈では、心理的診断
結論
NCCAHは、20-50%の酵素活性の保持のために、CAHのより頻繁かつ軽度の形態と考えられている。 ほとんどの患者は男性ホルモンの超過分と関連している徴候による彼らの生命のあらゆる段階で医師の診察を追求するかもしれません。 これらは早期のpubarche、急速な成長、多毛症、アクネ、menstrual不規則性、または不妊、また他の多くのより少なく顕著な明示を含んでいます。 良好な初期スクリーニング試験としての17OHPの早朝ベースライン値とACTH刺激によるさらなる評価、および境界結果の場合には遺伝子検査が推奨される。 原則として、androstenedioneおよびない17OHPのレベルはフォローアップの間に使用されるべきです。
治療の決定は事実の評価に基づいて行われ、個別化されたアプローチに従うべきである。 処置は患者が徴候、例えば、陰毛および体毛の早い手始めそして急速な進行、急速な成長、および/または骨格進歩の子供、またはoligomenorrhea、アクネ、hirsutism、不妊、またはこれら 遺伝の勧めることは強く父のgenotypingと同様、想像したいNCCAHの女性で助言されます。 患者の全体的な管理には、グルココルチコイド療法の可能性のある合併症または疾患の代謝関連症状の管理がさらに含まれる。 最後に、これらの患者の適切な管理に関連する多くの治療上の問題がまだ解明されていないことを考えると、主治医はこの分野のすべての発展を最新に保ち、新しいデータを彼の臨床実践に統合することが非常に重要です。 確かに、この分野でのさらなる臨床研究が不可欠です。
要約すると、NCCAH女性のために、理想的なアプローチは、彼女が彼女の人生を通して彼女に同行する症状指向の治療と一緒に、小児科から大人の内分泌 Hyperandrogenicシステムに影響を与える多数の要因を与えられて彼らの食餌療法の習慣を改善し、練習を高め、そして重量の軽減を目指すことによってより健康 さらに、特定のケースでは、心理的支援はしばしば有益である。 さらに、皮膚科医、婦人科医、心理学者など、この障害の治療に関与する分野の専門家は、NCCAHの疑わしいまたは既に診断された症例の管理について深い理 結論として、私たちは心の中でアメリカの天文学者ヴェラ*クーパー*ルービンの洞察力の引用を維持してみましょう:”観測は私たちの先入観を変更するために私たちを強制するとき、科学は最高の進歩します。”
著者の貢献
リストされているすべての著者は、仕事に実質的な、直接的かつ知的な貢献をし、出版のためにそれを承認しました。
利益相反に関する声明
著者らは、この研究は、利益相反の可能性と解釈される可能性のある商業的または財政的関係がない場合に行われたと宣言している。
1. Speiser PW、白のPC。 先天性副腎過形成。 N Engl J Med. (2003) 349:776–88. doi:10.1056/Nejmra021561
PubMed Abstract|CrossRef Full Text/Google Scholar
2. トゥルクAF,Auchus RJ. Adrenal steroidogenesis and congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. (2015) 44:275–96. doi: 10.1016/j.ecl.2015.02.002
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Speiser PW. Nonclassic adrenal hyperplasia. Rev Endocr Metab Disord. (2009) 10:77–82. doi: 10.1007/s11154-008-9097-x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Gidlöf S,Falhammar H,Thilén A,von Döbeln U,Ritzén M,Wedell A,et al. スウェーデンにおける先天性副腎過形成の百年:レトロスペクティブ、人口ベースのコホート研究。 ランセット糖尿病内分泌。 (2013) 1:35–42. doi:10.1016/S2213-8587(13)70007-X
PubMed Abstract|CrossRef Full Text/Google Scholar
5. Speiser PW、デュポンB、ルビンシュタインP、広場A、カステランA、新しいMI。 非古典的なステロイド21-ヒドロキシラーゼ欠乏症の頻度が高い。 Am J Hum Genet. (1985) 37:650–67.
PubMed Abstract | Google Scholar
6. Wilson RC, Nimkarn S, Dumic M, Obeid J, Azar MR, Azar M, et al. Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Mol Genet Metab. (2007) 90:414–21. doi: 10.1016/j.ymgme.2006.12.005
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Miller WL. Clinical review 54: genetics, diagnosis, and management of 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1994) 78:241–6. doi: 10.1210/jcem.78.2.8106606
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Witchel SF, Azziz R. Nonclassic congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2010) 2010:625105. doi: 10.1186/1687-9856-2010-625105
PubMed Abstract | CrossRef Full Text | Google Scholar
9. ウィルソンRC、メルカドAB、チェンKC、新しいMI。 ステロイド21-ヒドロキシラーゼ欠乏症:遺伝子型は表現型を予測しない可能性があります。 Jクリノールメタブ… (1995) 80:2322–9. 土井:10.1210/jc.80.8.2322
CrossRef全文|Google Scholar
10. 白いパソコン。 先天性副腎過形成のための新生児スクリーニング。 ナターシャ-レヴリノールズィー。 (2009) 5:490–8. 土井:10.1038/nrendo.2009.148
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Kohn B, Levine LS, Pollack MS, Pang S, Lorenzen F, Levy D, et al. Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. (1982) 55:817–27. doi: 10.1210/jcem-55-5-817
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Moran C,Azziz R,Carmina E,Dewailly D,Fruzzetti F,Ibañez L,et al. 21-ヒドロキシラーゼ欠損非古典的な副腎過形成は進行性疾患である:多施設研究。 Am J産科婦人科医。 (2000) 183:1468–74. 土井:10.1067/mob.2000.108020
PubMed Abstract|CrossRef Full Text|Google Scholar
13。 Azziz R,Hincapie LA,Knochenhauer ES,Dewailly D,Fox L,Boots LR. 高アンドロゲン女性の間で21-ヒドロキシラーゼ欠損非古典的な副腎過形成のためのスクリーニング:前向き研究。 フェチルステリル… (1999) 72:915–25. doi:10.1016/S0015-0282(99)00383-0
PubMed Abstract|CrossRef Full Text/Google Scholar
14。 Livadas S,Dracopoulou M,Dastamani A,Sertedaki A,Maniati-Christdi M,Magiakou A-M,et al. CYP21A2遺伝子の突然変異によって引き起こされる非古典的な生来の副腎の増殖の280人の個人の臨床、ホルモン性および分子調査結果のスペクトル。 クリニーク(Oxf)とも呼ばれる。 (2015) 82:543–9. 土井:10.1111/cen.12543
PubMed Abstract|CrossRef Full Text|Google Scholar
15。 Bidet M,Bellanné-Chantelot C,Galand-Portier M-B,Tardy V,Billaud L,Laborde K,et al. 161非古典的な先天性副腎過形成21-ヒドロキシラーゼ欠乏症と330家族のメンバーによるコホートの臨床および分子特性評価。 Jクリノールメタブ… (2009) 94:1570–8. 土井:10.1210/jc.2008-1582
PubMed Abstract|CrossRef Full Text|Google Scholar
16。 Escobar-Morreale HF,Sanchón R,San Millán JL. Hyperandrogenic徴候および印と示す女性間のnonclassical生来の副腎の増殖の有病率の前向き研究。 Jクリノールメタブ… (2008) 93:527–33. 土井:10.1210/jc.2007-2053
PubMed Abstract|CrossRef Full Text|Google Scholar
17。 Dewailly D,Vantyghem-Haudiquet MC,Sainsard C,Buvat J,Cappoen JP,Ardaens K,et al. 後期発症21-ヒドロキシラーゼ欠乏症における臨床的および生物学的表現型。 Jクリノールメタブ… (1986) 63:418–23. 土井:10.1210/jcem-63-2-418
PubMed Abstract|CrossRef Full Text/Google Scholar
18。 Gönç EN,Ozön ZA,Alikaşifoglu A,Engiz O,Bulum B,Kandemir N.(2011). 基礎血清17-OHプロゲステロンは、早期副腎における非古典的な先天性副腎過形成を予測するための信頼できるパラメータですか? トゥルク-ジャイルズ(Turk Joyz)。 53:274–80.
PubMed Abstract/Google Scholar
19。 Speiser PW,Azziz R,Baskin LS,Ghizzoni L,Hensle TW,Merke DP,et al. ステロイド21-ヒドロキシラーゼ欠乏症による先天性副腎過形成:内分泌学会臨床診療ガイドライン。 Jクリノールメタブ… (2010) 95:4133–60. 土井:10.1210/jc.2009-2631
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Dacou-Voutetakis C, Dracopoulou M. High incidence of molecular defects of the CYP21 gene in patients with premature adrenarche. J Clin Endocrinol Metab. (1999) 84:1570–4. doi: 10.1210/jc.84.5.1570
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Ambroziak U,Kepczynska-Events I,Kurylowicz,Małunowicz EM,Wójcicka,Miskiewicz P,et al. 血清基底またはACTH刺激後の17-ヒドロキシプロゲステロンに基づく21-ヒドロキシラーゼ欠乏症による副腎皮質の非古典的な先天性過形成の診断は、偽陽性診断につながる可能性がある。 クリニーク(Oxf)とも呼ばれる。 (2016) 84:23–9. doi:10.1111/price12935
PubMed Abstract|CrossRef Full Text|Google Scholar
22。 Ambroziak Y,Kepczynska-Events I,Kurylowicz,Wysłouch-Cieszyńska A,Małunowicz EM,Bartoszewicz,et al. LC-MS/MSは21-ヒドロキシラーゼの不足の方のスクリーニングを改善します。 ジノールエンデコリノール… (2015) 31:296–300. doi:10.3109/09513590.2014.994599
PubMed Abstract|CrossRef Full Text|Google Scholar
23。 bbc,CC,ゴールドマンMM,張K,クラーク-N,リッツ再ウェルトCK. 液体クロマトグラフィー-タンデム質量分析を用いた多嚢胞性卵巣症候群および対照女性におけるサーティーンステロイドホルモンの同時測定。 プロスワン ら(2 0 1 4)9:e9 3 8 0 5. doi:10.1371/ジャーナル。ポネ0093805
PubMed Abstract|CrossRef Full Text|Google Scholar
24。 Ray JA、Kushnir MM、Yost RA、Rockwood AL、Wayne Meikle a.差動イオン移動度分析と組み合わせたLC-MS/MSによる5つの内因性ステロイドの測定における性能向上。 クリン-チム-アクタ (2015) 438:330–6. 土井:10.1016/j.cca.2014.07.036
PubMed Abstract|CrossRef Full Text|Google Scholar
25。 Stoupa A,González-Briceño L,Pinto G,Samara-Boustani D,Thalassinos C,Flechtner I,et al. 非古典的な先天性副腎過形成におけるテトラコサクタイド(Synacthen®)試験に対する不十分なコルチゾール応答:ルールの例外? ホルム-レ-パエディアトル… (2015) 83:262–7. 土井:10.1159/000369901
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. (1992) 90:584–95. doi: 10.1172/JCI115897
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Temeck JW,Pang SY,Nelson C,New MI. 早期pubarcheにおけるステロイド形成の遺伝的欠陥。 Jクリノールメタブ… (1987) 64:609–17. 土井:10.1210/jcem-64-3-609
PubMed Abstract|CrossRef Full Text/Google Scholar
28。 Voutilainen R,Jääskeläinen J.早期adrenarche:病因,臨床所見,および結果. Jステロイドバイオケムモールバイオール… (2015) 145:226–36. 土井:10.1016/j.jsbmb.2014.06.004
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Oberfield SE, Sopher AB, Gerken AT. Approach to the girl with early onset of pubic hair. J Clin Endocrinol Metab. (2011) 96:1610–22. doi: 10.1210/jc.2011-0225
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Janus D, Wojcik M, Malunowicz E, Starzyk JB. 15ヶ月の子供における再発性唇癒着のケースは、21-ヒドロキシラーゼ欠乏症による無症候性の非古典的な先天性副腎過形成を有する。 J-PediatrのEndocrinolのMetab。 (2012) 25:1017–21. doi:10.1515/jpem-2012-0190
PubMed Abstract|CrossRef Full Text/Google Scholar
31。 Gopal-Kothandapani JS、Petkar A、O’Shea E、Banerjee i.非古典的な先天性副腎過形成の異常な提示としての肛門周囲の髪。 (2013)2013:bcr2013010123. 土井:10.1136/bcr-2013-010123
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Falhammar H, Nordenström A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine. (2015) 50:32–50. doi: 10.1007/s12020-015-0656-0
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Chrousos GP, Loriaux DL, Mann DL, Cutler GB. Late-onset 21-hydroxylase deficiency mimicking idiopathic hirsutism or polycystic ovarian disease. Ann Intern Med. (1982) 96:143–8. doi: 10.7326/0003-4819-96-2-143
CrossRef Full Text | Google Scholar
34. Witchel SF. Non-classic congenital adrenal hyperplasia. Steroids. (2013) 78:747–50. doi: 10.1016/j.steroids.2013.04.010
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Carmina E, Rosato F, Jannì A, Rizzo M, Longo RA. Extensive clinical experience: relative prevalence of different androgen excess disorders in 950 women referred because of clinical hyperandrogenism. J Clin Endocrinol Metab. (2006) 91:2–6. doi: 10.1210/jc.2005-1457
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Moran C,Azziz R,Weintrob N,Witchel SF,Rohmer V,Dewailly D,et al. 21-ヒドロキシラーゼ欠損非古典的副腎過形成を有する女性の生殖転帰。 Jクリノールメタブ… (2006) 91:3451–6. 土井:10.1210/jc.2006-0062
PubMed Abstract|CrossRef Full Text|Google Scholar
37。 レビンJH、カルミナE、ロボRA。 多嚢胞性卵巣症候群の患者の不適切な性腺刺激ホルモン分泌は、成人発症の先天性副腎過形成の患者のそれと同様であるか? フェチルステリル… (1991) 56:635–40. doi: 10.1016/S0015-0282(16)54592-0
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Carmina E, Lobo RA. Ovarian suppression reduces clinical and endocrine expression of late-onset congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Fertil Steril. (1994) 62:738–43. doi: 10.1016/S0015-0282(16)56998-2
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Ludwig E.女性の性別で起こるアンドロゲン性脱毛症(一般的な脱毛症)のタイプの分類。 Br J Dermatol. (1977) 97:247–54. ドイ:10.1111/j.1365-2133.1977.15179x
PubMed Abstract|CrossRef Full Text|Google Scholar
40。 Trakakis E,Papadavid E,Dalamaga M,Koumaki D,Stavrianeas N,Rigopoulos D,et al. にきびを持つギリシャの女性における21-ヒドロキシラーゼ欠乏症による非古典的な先天性副腎過形成の有病率:病院ベースの断面研究。 J Eur Acad Dermatol Venereol. (2013) 27:1448–51. ドイ:10.1111/j.1468-3083.2012.04613.x
PubMed Abstract|CrossRef Full Text/Google Scholar
41。 Arlt W,Willis DS,Wild SH,Krone N,Doherty EJ,Hahner S,et al. 先天性副腎過形成を有する成人の健康状態:203人の患者のコホート研究。 Jクリノールメタブ… (2010) 95:5110–21. 土井:10.1210/jc.2010-0917
PubMed Abstract|CrossRef Full Text|Google Scholar
42。 Saygili F、Oge A、Yilmaz C.21-ヒドロキシラーゼ欠乏症による非古典的な先天性副腎過形成を有する女性における高インスリン血症およびインスリン非感受性:血清レプチンレベルと慢性高インスリン血症との関係。 Horm Res.(2 0 0 5)6 3:2 7 0−4. 土井:10.1159/000086363
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Speiser PW, Serrat J, New MI, Gertner JM. Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1992) 75:1421–4. doi: 10.1210/jc.75.6.1421
PubMed Abstract | CrossRef Full Text | Google Scholar
44. Han TS,Stimson RH,Rees DA,Krone N,Willis DS,Conway GS,et al. 先天性副腎過形成を有する成人におけるグルココルチコイド治療レジメンおよび健康転帰。 クリニーク-エンデクリノールトランスミッション (2013) 78:197–203. 土井:10.1111/cen.12045
PubMed Abstract|CrossRef Full Text|Google Scholar
45。 Van Staa TP、Leufkens HG、Abenhaim L、Zhang B、Cooper C.経口コルチコステロイドの使用および骨折のリスク。 J Bone Miner Res.(2 0 0 0)1 5:9 9 3−1 0 0 0. ドイ:10.1359/jbmr.2000.15.6.993
PubMed Abstract | CrossRef Full Text | Google Scholar
46. Nebesio TD, Renbarger JL, Nabhan ZM, Ross SE, Slaven JE, Li L, et al. Differential effects of hydrocortisone, prednisone, and dexamethasone on hormonal and pharmacokinetic profiles: a pilot study in children with congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2016) 2016:17. doi: 10.1186/13633-016-0035-5
PubMed Abstract|CrossRef Full Text/Google Scholar
47。 Weintrob N、Dickerman Z、Sprecher E、Galatzer A、Pertzelan A.幼児期および小児期における非古典的な21-ヒドロキシラーゼ欠乏症:思春期および最終的な高さに対する治療開始時間の影響。 ユール-ジュリエット(著), (1997) 136:188–95. 土井:10.1530/eje.0.1360188
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Bonfig W, Bechtold S, Schmidt H, Knorr D, Schwarz HP. Reduced final height outcome in congenital adrenal hyperplasia under prednisone treatment: deceleration of growth velocity during puberty. J Clin Endocrinol Metab. (2007) 92:1635–9. doi: 10.1210/jc.2006-2109
PubMed Abstract | CrossRef Full Text | Google Scholar
49. Dörr HG. 21-ヒドロキシラーゼ欠乏症による古典的な先天性副腎過形成患者の成長。 Horm Res.(2007)68(Suppl5):93-9. doi:10.1159/000110587
PubMed Abstract|CrossRef Full Text|Google Scholar
50。 Hoepffner W、Kaufhold A、Willgerodt H、Keller E.21-ヒドロキシラーゼ欠乏症による古典的な先天性副腎過形成の患者は、目標の高さを達成することができます:ライプツィヒの経験。 Horm Res.(2008)70:42-50. 土井:10.1159/000129677
PubMed Abstract | CrossRef Full Text | Google Scholar
51. van der Kaay D, van den Akker E. Ultralow-dose dexamethasone to preserve endogenous cortisol stress response in nonclassical congenital adrenal hyperplasia: a new promising treatment. Int J Endocrinol Metab. (2014) 12:e14657. doi: 10.5812/ijem.14657
CrossRef Full Text | Google Scholar
52. Lin-Su K, Vogiatzi MG, Marshall I, Harbison MD, Macapagal MC, Betensky B, et al. Treatment with growth hormone and luteinizing hormone releasing hormone analog improves final adult height in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. (2005) 90:3318–25. doi: 10.1210/jc.2004-2128
PubMed Abstract | CrossRef Full Text | Google Scholar
53. McCann-Crosby B, Chen M-J, Lyons SK, Lin Y, Axelrad M, Dietrich JE, et al. Nonclassical congenital adrenal hyperplasia: 治療と移行のターゲット。 Pediatr Endocrinol Rev.(2014)12:224-38.
PubMed Abstract/Google Scholar
54。 Ogilvie CM,クラウチNS,ラムスビー G,クレイトンSM,遼L-M,コンウェイGS. 成人における先天性副腎過形成:医学的、外科的および心理的問題のレビュー。 クリニーク-エンデクリノールトランスミッション (2006) 64:2–11. ドイ:10.1111/j.1365-2265.2005.02410.x
PubMed Abstract | CrossRef Full Text | Google Scholar
55. Mah PM, Jenkins RC, Rostami-Hodjegan A, Newell-Price J, Doane A, Ibbotson V, et al. Weight-related dosing, timing and monitoring hydrocortisone replacement therapy in patients with adrenal insufficiency. Clin Endocrinol. (2004) 61:367–75. doi: 10.1111/j.1365-2265.2004.02106.x
PubMed Abstract | CrossRef Full Text | Google Scholar
56. ディアスA,A上のPCのLaufer氏逆子LLAA,AH上のOとGCのケアAC. 少女および青年の月経:月経周期をバイタルサインとして使用する。 小児科… (2006) 118:2245–50. ドイ:10.1542/peds.2006-2481
CrossRef全文|Google Scholar
57。 メルケDP、ポパスDP。 先天性副腎過形成を有する青年の管理。 ランセット糖尿病内分泌。 (2013) 1:341–52. 土井:10.1016/S2213-8587(13)70138-4
PubMed Abstract | CrossRef Full Text | Google Scholar
58. Verma S, Green-Golan L, VanRyzin C, Drinkard B, Mehta SP, Weise M, et al. Adrenomedullary function in patients with nonclassic congenital adrenal hyperplasia. Horm Metab Res. (2010) 42:607–12. doi: 10.1055/s-0030-1253385
PubMed Abstract | CrossRef Full Text | Google Scholar
59. New MI. Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2006) 91:4205–14. doi: 10.1210/jc.2006-1645
PubMed Abstract | CrossRef Full Text | Google Scholar
60. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2018) 103:4043–88. doi: 10.1210/jc.2018-01865
PubMed Abstract | CrossRef Full Text | Google Scholar
61. Kruse B, Riepe FG, Krone N, Bosinski HAG, Kloehn S, Partsch CJ, et al. Congenital adrenal hyperplasia – how to improve the transition from adolescence to adult life. Exp Clin Endocrinol Diabetes. (2004) 112:343–55. doi: 10.1055/s-2004-821013
PubMed Abstract | CrossRef Full Text | Google Scholar
62. Gastaud F,Bouvattier C,Duranteau L,Brauner R,Thibaud E,Kutten F,et al. 先天性副腎過形成の古典的な形態を有する女性における性的および生殖的転帰の障害。 Jクリノールメタブ… (2007) 92:1391–6. 土井:10.1210/jc.2006-1757
PubMed Abstract|CrossRef Full Text|Google Scholar
63。 Mercè Fernández-Balsells M,Muthusamy K,Smushkin G,Lampropulos JF,Elamin MB,Abu Elnour NO,et al. 21ヒドロキシラーゼ(CYP21A2)の不足のために古典的な生来の副腎の増殖の危険がある状態の妊娠のvirilizationの防止のための出生前のdexamethasoneの使用:系統検討およ クリニーク-エンデクリノールトランスミッション (2010) 73:436–44. ドイ:10.1111/j.1365-2265.2010.03826.x
CrossRef全文|Google Scholar
64。 Bidet M,Bellanné-Chantelot C,Galand-Portier MB,Golmard JL,Tardy V,Morel Y,et al. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2010) 95:1182–90. doi: 10.1210/jc.2009-1383
PubMed Abstract | CrossRef Full Text | Google Scholar
65. Birnbaum MD, Rose LI. Late onset adrenocortical hydroxylase deficiencies associated with menstrual dysfunction. Obstet Gynecol. (1984) 63:445–51.
PubMed Abstract | Google Scholar
66. Cuhaci N, Aydin C, Yesilyurt A, Pinarli FA, Ersoy R, Cakir B. Nonclassical congenital adrenal hyperplasia and pregnancy. Case Rep Endocrinol. (2015) 2015:296924. doi: 10.1155/2015/296924
PubMed Abstract | CrossRef Full Text | Google Scholar
67. Deneux C, Tardy V, Anne D, Mornet E, Billaud L, Charron D, et al. 21-ヒドロキシラーゼ欠乏症による非古典的な先天性副腎過形成を有する56人の女性における表現型-遺伝子型相関。 Jクリノールメタブ… (2001) 86:207–13. 土井:10.1210/jcem.86.1.7131
PubMed Abstract|CrossRef Full Text|Google Scholar
68。 Simonetti L,Bruque CD,Fernández CS,Benavides-Mori B,Delea M,Kolomenski JE,et al. CYP21A2突然変異の更新:データベースおよび出版された遺伝の変形の広範囲の分析。 フム-ムタット (2018) 39:5–22. 土井:10.1002/humu.23351
PubMed Abstract | CrossRef Full Text | Google Scholar
69. OMIM Entry – * 613815 – CYTOCHROME P450 FAMILY 21 SUBFAMILY A POLYPEPTIDE 2; CYP21A2.
Google Scholar
70. Miller WL, Witchel SF. Prenatal treatment of congenital adrenal hyperplasia: risks outweigh benefits. Am J Obstet Gynecol. (2013) 208:354–9. doi: 10.1016/j.ajog.2012.10.885
PubMed Abstract | CrossRef Full Text | Google Scholar
71. Lekarev O, New MI. Adrenal disease in pregnancy. Best Pract Res Clin Endocrinol Metab. (2011) 25:959–73. doi: 10.1016/j.beem.2011.08.004
PubMed Abstract | CrossRef Full Text | Google Scholar
72. New MI, Abraham M, Yuen T, Lekarev O. An update on prenatal diagnosis and treatment of congenital adrenal hyperplasia. Semin Reprod Med. (2012) 30:396–9. doi: 10.1055/s-0032-1324723
PubMed Abstract | CrossRef Full Text | Google Scholar
73. Kazmi D, Bailey J, Yau M, Abu-Amer W, Kumar A, Low M, et al. New developments in prenatal diagnosis of congenital adrenal hyperplasia. J Steroid Biochem Mol Biol. (2017) 165:121–3. doi: 10.1016/j.jsbmb.2016.06.016
PubMed Abstract | CrossRef Full Text | Google Scholar
74. Clayton PE,Miller WL,Oberfield SE,Ritzén EM,Sippell WG,Speiser PW,et al. 小児科の内分泌学およびローソンwilkinsの小児科の内分泌の社会のためのヨーロッパの社会からの21-hydroxylaseの不足の一致の声明。 Horm Res.(2 0 0 2)5 8:1 8 8−9 5. doi:10.1159/000065490
PubMed Abstract|CrossRef Full Text|Google Scholar
75。 Nandagopal R,Sinaii N,Avila NA,Van Ryzin C,Chen W,Finkielstain GP,et al. 不可解な非古典的な先天性副腎過形成を有する親の表現型プロファイリング:145の無関係な家族における所見。 ユール-ジュリエット(著), (2011) 164:977–84. doi:10.1530/EJE-11-0019
PubMed Abstract|CrossRef Full Text/Google Scholar
76。 高須N、Nakachi K、比嘉H.グレーブスの甲状腺機能亢進症の開発は、以前に認識されていない非古典的な21-ヒドロキシラーゼ欠乏症の患者で副腎の危機を引き起こ インターンメッド (2010) 49:1395–400. 土井:10.2169/内科.49.3573
PubMed Abstract | CrossRef Full Text | Google Scholar
77. Meyer-Bahlburg HFL, Dolezal C, Baker SW, Carlson AD, Obeid JS, New MI. Prenatal androgenization affects gender-related behavior but not gender identity in 5-12-year-old girls with congenital adrenal hyperplasia. Arch Sex Behav. (2004) 33:97–104. doi: 10.1023/B:ASEB.0000014324.25718.51
CrossRef Full Text | Google Scholar
78. Meyer-Bahlburg HFL、Dolezal C、Baker SW、New MI。 出生前アンドロゲン過剰の程度の関数としての古典的または非古典的な先天性副腎過形成を有する女性の性的指向。 アーチ性行動。 (2008) 37:85–99. 土井:10.1007/10508-007-9265-1P>
PubMed Abstract|CrossRef Full Text|Google Scholar