La sclera blu è causata dalla magrezza e dalla trasparenza delle fibre di collagene della sclera che consentono la visualizzazione dell’uvea sottostante (Figura 1). La sclera è il mantello esterno bianco dell’occhio, che circonda l’iride può essere assottigliato in malattie congenite come l’osteogenesi imperfetta 1). Altre malattie associate alla sclera blu includono disturbi multipli del tessuto connettivo, come la sindrome della cornea fragile 2), la sindrome di Marshall e Stickler 3), POESIE (polineuropatia, organomegalia, endocrinopatia, proteine monoclonali e cambiamenti cutanei) sindrome 4), sindrome di Marfan, sindrome di Ehlers–Danlos, pseudoxanthoma elasticum e sindrome di Willems De Vries, per citarne alcuni. Disturbi ossei e del sangue anche sulla lista includono anemia Diamond–Blackfan, grave anemia da carenza di ferro, la malattia di Paget giovanile, e la carenza di fosfatasi acida 5).
Le forme gravi di osteogenesi imperfetta sono spesso diagnosticate precocemente, ma i casi lievi possono essere notati solo più tardi nella vita. Il colore blu-grigio della sclera è dovuto alle vene coroidali sottostanti che mostrano attraverso. Ciò è dovuto al fatto che la sclera è più sottile del normale perché il collagene di tipo I difettoso non si sta formando correttamente 6). La sclera è una struttura densa del tessuto connettivo poco vascolarizzata composta da collagene di tipo I, III, IV, V, VI e VIII, nonché elastina, proteoglicani e glicoproteine 7).
Negli Stati Uniti, l’incidenza di osteogenesi imperfetta è stimata essere una ogni 20.000 nati vivi. Si stima che 20.000-50.000 persone siano affette da osteogenesi imperfetta negli Stati Uniti 8).
Le persone con osteogenesi imperfetta nascono con tessuto connettivo difettoso, o senza la capacità di farlo, di solito a causa di una carenza di collagene di tipo I. Questa carenza deriva da una sostituzione aminoacidica della glicina in aminoacidi più ingombranti nella struttura a tripla elica del collagene. Di conseguenza, il corpo può rispondere idrolizzando la struttura impropria del collagene 9). Se il corpo non distrugge il collagene improprio; la relazione tra le fibrille di collagene e i cristalli di idrossiapatite per formare l’osso viene alterata, causando fragilità. Come una malattia genetica, osteogenesi imperfecta è stato storicamente visto come una malattia autosomica dominante di collagene di tipo I. La maggior parte dei casi è stata causata da mutazioni nei geni COL1A1 e COL1A2 10). Negli ultimi anni, c’è stata l’identificazione di forme autosomiche recessive. Almeno sette sottoinsiemi sono stati descritti anche se quattro sottotipi principali sono più comuni e vanno da lieve a grave. Gli individui con osteogenesi imperfetta di tipo I hanno una piccola deformità ossea, una sclera blu persistente, un’altezza quasi normale entro l’età adulta e una probabilità di perdita dell’udito >del 50% entro l’età adulta. I pazienti con osteogenesi imperfetta letale perinatale (Tipo II) mostrano la massima gravità, con fratture multiple in utero o dal parto. Questi pazienti di solito sono nati morti o muoiono precocemente. La gravità dell’osteogenesi imperfetta dipende dal difetto genetico specifico. La maggior parte dei casi di osteogenesi imperfetta sono ereditati da un genitore. Tuttavia, alcuni casi sono il risultato di nuove mutazioni genetiche. Una persona con osteogenesi imperfetta ha una probabilità del 50% di trasmettere il gene e la malattia ai propri figli 11).
Figura 1. Anatomia dell’occhio
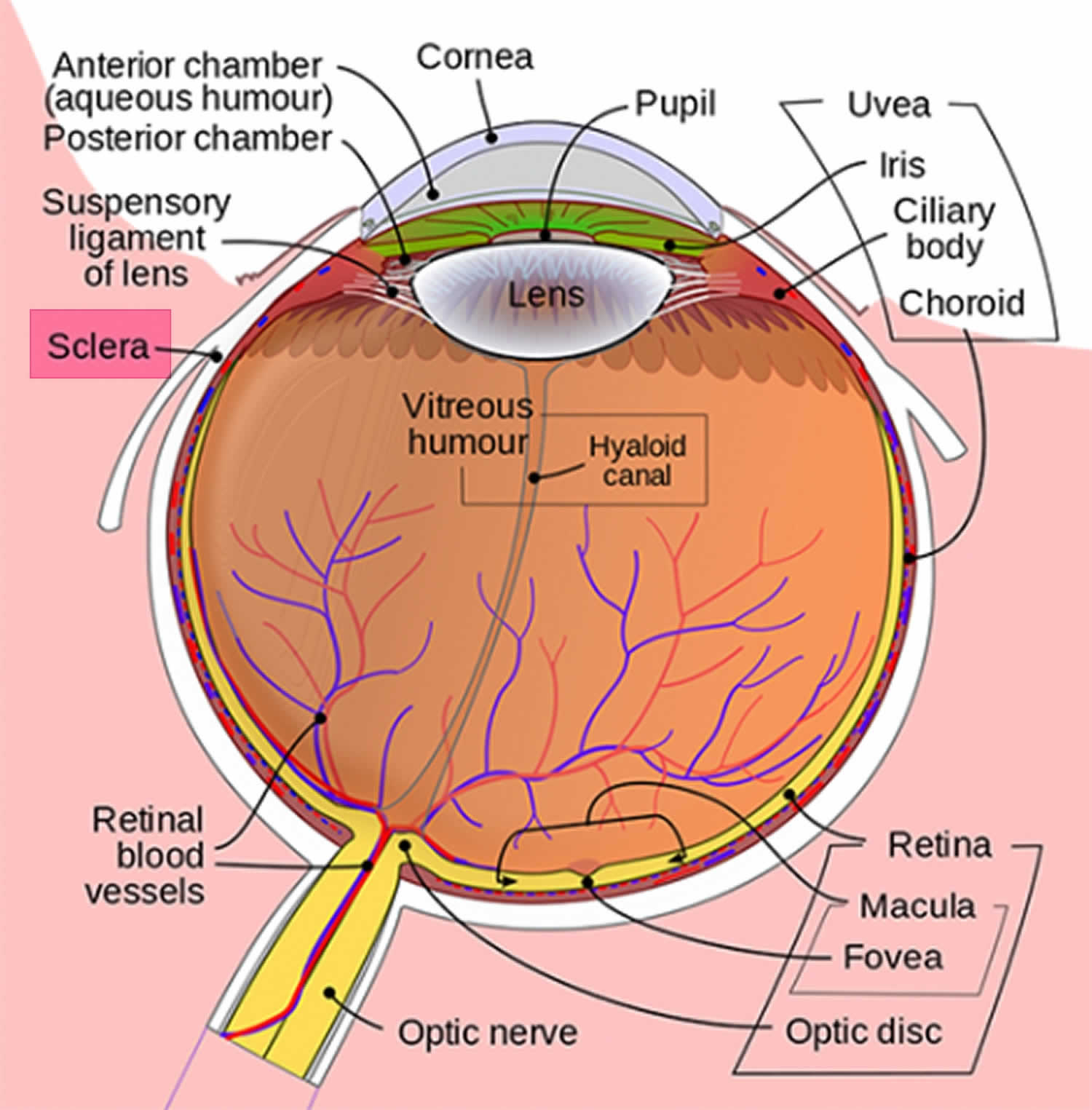
Figura 2. Sclera blu
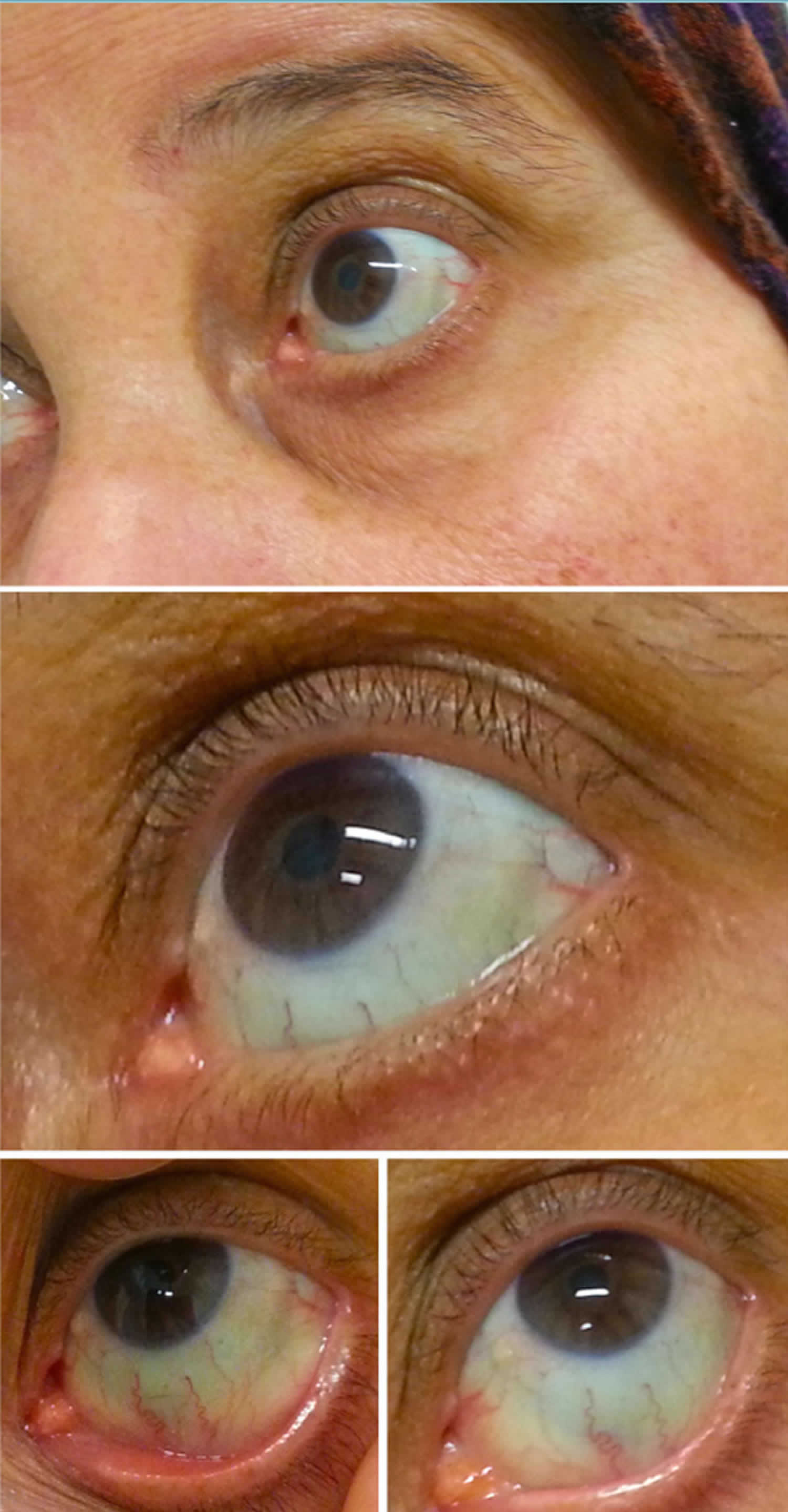
La sclera blu causa
La sclera blu è la manifestazione più consistente dell’osteogenesi imperfetta, che deriva da una mutazione in COLIA1 e COL1A2, che codifica per il procollagene di tipo I. Tuttavia, le classificazioni di questa condizione (tipi IV-VI) sono state identificate con sclerae normali. L’osteogenesi imperfetta è anche associata a fragilità anormale delle ossa e sordità.
Cornea fragile, sclera blu e capelli rossi sono associati alla sindrome della cornea fragile, una condizione che presenta anche anomalie scheletriche,dentali e cutanee 13). Una mutazione missense in ZNF469 è stata trovata per essere causativa per la malattia.
Altre condizioni di base associate alla formazione di sclera blu includono la sindrome di Ehlers-Danlos (tipo VI), pseudoxanthoma elasticum, cornea plana, sclerocornea periferica, buftalmo, cheratocono, cheratoglobus, alta miopia, stafiloma ciliare/equatoriale, melanocitosi oculodermica e microcornea. Raramente, la sclera blu si verifica con sindrome di Hallermann-Streiff, sindrome di Marfan, sindrome di Turner, sindrome di Cheney, sindrome di Menkes, pyknodysostosis, cornee fragili o displasia ectodermica.
La sclera blu può verificarsi anche nei neonati normali durante i primi mesi di vita; tuttavia, la persistenza di una decolorazione blu nel tempo può suggerire la presenza di una pressione intraoculare elevata. I neonati prematuri mostrano spesso sclerae blu, in particolare quelli di origine caucasica.
La sclera blu può anche verificarsi in isolamento come anomalia autosomica dominante o autosomica recessiva ereditaria 14).
Sintomi della sclera blu
La sclera blu si presenta con un aspetto bluastro alla sclera e può essere associata a causa patologica o non patologica. Altre caratteristiche oculari dei disturbi del tessuto connettivo associati alla sclera blu includono cornea sottile, piega epicantale, miopia, cheratocono e striature angioidi. Le caratteristiche sistemiche dei disturbi del tessuto connettivo associati alla sclera blu includono anomalie cutanee, anomalie cardiache, cifoscoliosi, ipermobilità articolare, ossa fragili, anomalie dell’udito, anomalie vascolari e anomalie gastrointestinali 15).
Diagnosi della sclera blu
La valutazione diagnostica della sclera blu comporta l’esame esterno, la biomicroscopia della lampada a fessura e la valutazione sistemica per i disturbi associati.Sebbene non esista un test definitivo per l’osteogenesi imperfetta, il test genetico può confermare o escludere mutazioni note 16).
Trattamento della sclera blu
Il trattamento per la sclera blu comporta la diagnosi e il trattamento della causa sottostante. La sclera blu è principalmente dovuta a sindromi genetiche e, in misura minore, a disturbi non genetici e può verificarsi come effetto collaterale dell’assunzione di farmaci. Le sclerae blu sono comunemente associate a disturbi congeniti della sintesi del collagene, come l’osteogenesi imperfetta, la sindrome di Marfan, la sindrome di Ehlers–Danlos, lo pseudoxanthoma elasticum e la sindrome di Willems De Vries, per citarne alcuni. Anche i disturbi ossei e del sangue sono inclusi nell’elenco: anemia Diamond–Blackfan, grave anemia da carenza di ferro, malattia giovanile di Paget e carenza di fosfatasi acida 17). Le sclerae blu acquisite sono state descritte negli adulti con melanosi oculare, melanoma congiuntivale, alcaptonuria e malattia di Addison. Per le manifestazioni non oculari possono essere indicati appropriati rinvii a valutazioni pediatriche e ortopediche 18).
La consulenza genetica per disturbi ereditari associati può essere utile per i pazienti con sclerae altre manifestazioni di malattia sistemica.
L’intervento chirurgico può essere indicato in caso di diradamento estremo e perforazione. Il supporto strutturale può essere fornito da un innesto sclerale conservato o fascia lata utologa, in particolare nei casi che richiedono la sutura di un dispositivo come un impianto a tubo 19). Le indagini sistemiche e il trattamento devono essere considerati per affrontare la patologia sottostante.
Prognosi sclera blu
La prognosi varia con la presenza di manifestazioni oculari e sistemiche del disturbo sottostante. I pazienti con sclera blu sono a maggior rischio di rottura del globo o perforazione sclerale intraoperatoria durante la chirurgia oculare di routine. I disturbi sistemici sono anche prevalenti in pazienti come fistola carotide-cavernosa, rottura arteriosa, perdita dell’udito e fratture ossee 20).
Riferimenti