La struttura primaria di una proteina è definita come la sequenza di amminoacidi di cui è composta. Questa sequenza determina in ultima analisi la forma che la proteina adotta, in base alle limitazioni spaziali sulla disposizione degli atomi nella proteina, le proprietà chimiche dei residui di amminoacidi componenti e l’ambiente della proteina.
I legami peptidici che collegano i residui di amminoacidi in un polipeptide si formano in una reazione di condensazione tra il gruppo carbossilico acido di un amminoacido e il gruppo amminico basico di un altro amminoacido. Nel contesto di un peptide, il gruppo ammidico (CO–NH) viene indicato come gruppo peptidico.
Cruciale per una comprensione della struttura della proteina è una conoscenza della struttura del legame peptidico. Linus Pauling, nel 1930, ha usato la diffrazione dei raggi X per esaminare la natura del legame peptidico formato tra due amminoacidi. Ha riferito che il gruppo peptidico (CO–NH) ha una struttura planare rigida. Questa struttura è dovuta alle interazioni tra gli elettroni del doppio legame del gruppo carbonilico e quelli del legame C–N (Figura 2) tali che quest’ultimo acquisisce proprietà parziali (circa il 40%) di doppio legame.

Questo effetto è un esempio di risonanza che può essere pensato come una condivisione di elettroni tra legami. Poiché i singoli legami tra due atomi sono più lunghi dei doppi legami tra gli stessi due atomi, le lunghezze dei legami C–N e C=O nel gruppo peptidico differiscono da quelle osservate per questi legami in altri contesti in cui non si verifica la risonanza. Quindi il carattere parziale del doppio legame di C – N nel gruppo peptidico significa che questo legame è più breve di quanto sarebbe previsto per un legame singolo C–N, mentre il legame C=O, avendo un carattere di legame singolo parziale dovuto alla risonanza, è più lungo di quanto sarebbe previsto per un doppio legame C=O. Le lunghezze di legame nel gruppo peptidico sono indicate nella Figura 3. Confronta il legame C-N del gruppo peptidico con quello tra N e Ca (l’atomo C a cui sono attaccati il gruppo amminico e il gruppo carbossilico).

Esistono due possibili conformazioni del legame peptidico planare: nel gruppo trans peptidico, gli atomi di Ca si trovano sui lati opposti del legame peptidico (Figura 3a) e nel gruppo peptidico cis, gli atomi di Ca si trovano sullo stesso lato del legame peptidico (Figura 3b).
-
Considerando la disposizione spaziale e la vicinanza degli atomi nelle conformazioni cis e trans del legame peptidico, quale conformazione pensi sarebbe favorita?
-
La conformazione trans sarebbe energeticamente più favorevole della conformazione cis, poiché minimizza l’ostacolo sterico.
In generale, i legami peptidici sono nella conformazione trans. Tuttavia, forme cis possono verificarsi in legami peptidici che precedono un residuo di prolina. In questi casi, la forma cis è più stabile del solito poiché la catena laterale della prolina offre meno ostacoli. Tuttavia, i legami peptidici cis si verificano solo in circa il 10% dei casi di legami peptidici che precedono i residui di prolina.
Tenendo presente la natura planare del gruppo peptidico, si può vedere che una catena polipeptidica ha una spina dorsale costituita da una serie di gruppi peptidici planari rigidi collegati dagli atomi di Ca. La figura 4 mostra parte di un polipeptide con due gruppi peptidici planari nella conformazione trans. Si noti che sebbene la rotazione non sia consentita sui legami peptidici, esiste un potenziale di rotazione attorno ai legami Ca–N e Ca–C. Gli angoli di rotazione, chiamati angoli di torsione, su questi legami specificano la conformazione di una spina dorsale polipeptidica. Gli angoli di torsione attorno ai legami Ca-N e Ca-C sono indicati come ɸ (phi) e ψ. (psi), rispettivamente e sono definiti come 180° quando il polipeptide è nella conformazione planare estesa, come illustrato nella Figura 4.
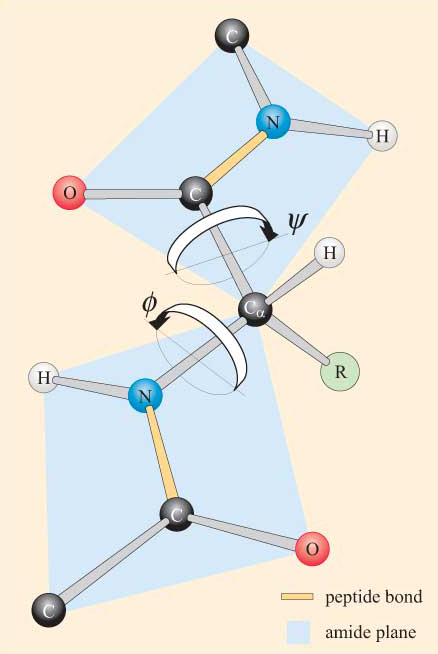
Non sarai sorpreso di apprendere che i vincoli sterici si applicano a ɸ e ψ.
Come risultato di questi vincoli sterici, solo alcuni valori di ɸ e ψ, e quindi conformazioni del peptide, sono consentiti mentre altri non lo sono.
È possibile calcolare questi valori consentiti per un determinato residuo nel contesto di un polipeptide. Questo calcolo viene eseguito determinando innanzitutto le distanze tra tutti gli atomi non legati in due gruppi peptidici vicini (come quelli in Figura 4) a tutti i possibili valori di ɸ e ψ. È più facilmente fatto per un polipeptide contenente solo un tipo di amminoacido. Una trama conformazionale di against contro against per un particolare residuo è nota come trama Ramachandran (dal suo inventore, G. N. Ramachandran). Tale trama permette di identificare quelle conformazioni (cioè per un particolare valore di ɸ e ψ) che sono stericamente favorevoli o sfavorevoli (come nella Figura 5), secondo i seguenti criteri:
-
Dove non c’è conflitto tra i raggi di van der Waals degli atomi non-bonding, una conformazione è “permessa”. Queste conformazioni si trovano nelle aree blu in Figura 5.
-
Le conformazioni che richiedono distanze interatomiche al limite di ciò che è ammissibile sono definite come conformazioni “limite esterno”. Si trovano nelle aree verdi in Figura 5.
-
Le conformazioni teoriche che richiedono due atomi non legati per essere più vicini l’uno all’altro rispetto ai raggi di van der Waals consentiti sono stericamente “proibite”. Questi si trovano nelle aree bianche in Figura 5.
Si noti che i valori di ɸ e ψ nella Figura 5 vanno da −180º a +180º. Girando il gruppo peptide attraverso 360º naturalmente lo riporterà alla sua posizione di partenza e-180º e +180º corrispondono alla stessa posizione. Pertanto la striscia verde nell’angolo in basso a sinistra del grafico in Figura 5 è contigua al campo nell’angolo in alto a sinistra.

-
Figura 5 per determinare se i seguenti valori di ɸ e ψ sono stericamente favorevole o sfavorevole: (a) ɸ = 90º e ψ = 90°; (b) ɸ = −90º e ψ = 90º.
-
(a) Sfavorevole; b) favorevole.
I diagrammi di Ramachandran possono essere costruiti per i polimeri di ciascuno dei 20 amminoacidi. È significativo notare che le trame di Ramachandran per molti residui di amminoacidi sono generalmente molto simili, avendo solo tre regioni con conformazioni favorevoli o tollerate (etichettate 1-3 nel grafico per la poli-l-alanina nella Figura 5). Le differenze si verificano, tuttavia. Ad esempio, dove la catena laterale (R in Figura 4) è ramificata vicino a Ca, come nel caso della treonina, occupa più spazio vicino alla spina dorsale del peptide e limita l’approccio degli atomi nei gruppi peptidici vicini. Di conseguenza, le conformazioni consentite (angles e angles angoli) sono più limitate per i polipeptidi degli amminoacidi ramificati.
-
La prolina è anche molto diversa dagli altri amminoacidi in termini di conformazioni consentite e solo per la poliprolina are sono tollerati valori da −85º a −35º. Pensando alla struttura della prolina, come puoi spiegare questo intervallo relativamente ristretto di valori permitted consentiti?
-
La catena laterale della prolina è legata covalentemente all’N del gruppo amminico, quindi in poliprolina, ci sarà meno libertà di rotazione sul legame Ca-N rispetto ad altri amminoacidi. Di conseguenza, i valori allowed consentiti saranno relativamente limitati rispetto ad altri amminoacidi.
-
La figura 6 mostra il grafico Ramachandran per i residui di glicina in una catena polipeptidica. Le regioni sono codificate a colori come nella Figura 5. Cosa puoi dire delle conformazioni che adotta la glicina? Considera la struttura della glicina. Perché la glicina differisce dagli altri residui per quanto riguarda le sue conformazioni?
-
La glicina ha una libertà conformazionale molto maggiore rispetto ad altri residui di aminoacidi, perché è meno ostacolata stericamente.

Le trame Ramachandran nelle figure 5 e 6 sono state generate rispettivamente per l-alanina e l-glicina sulla base delle distanze limite consentite ed esterne per i contatti interatomici, determinate da valori noti per i raggi di van der Waals degli atomi (Tabella 1).
Tabella 1 Distanze di Van der Waals per contatti interatomici.
| Tipo di contatto | Normalmente consentito/Å | Limite esterno/Å | |
|---|---|---|---|
| H···H | 2.0 | 1.9 |
3.0 |
Sono quindi predittivi piuttosto che veri e propri grafici conformazionali. Possiamo, naturalmente, utilizzare la diffrazione dei raggi X per determinare sperimentalmente i valori “reali” di residues e residues per i residui in un polipeptide. Nella figura 7, i valori ɸ e ψ per tutti i residui (ad eccezione della glicina e della prolina) in un certo numero di strutture diverse sono stati determinati mediante diffrazione a raggi X ad alta risoluzione e tracciati su un diagramma di Ramachandran. Possiamo vedere che c’è una corrispondenza sorprendente tra conformazioni previste e reali. Si noti, tuttavia, che ci sono alcuni residui la cui conformazione mappa alle aree ‘proibite’. La maggior parte di questi residui mappa nella regione tra le regioni “consentite” 2 e 3, intorno a ψ = 0.

-
Guarda di nuovo la Figura 4 e immagina di poter ruotare il gruppo peptidico più in alto di 180° in modo che ψ = 0. Quali gruppi sono suscettibili di conflitto in questa conformazione?
-
I gruppi NH di gruppi peptidici adiacenti entreranno in conflitto tra loro, essendo costretti in prossimità.
Il conflitto associato a queste conformazioni può essere sistemato da un piccolo grado di torsione del legame peptidico. Così, in tali conformazioni il gruppo peptide è attorcigliato dalla sua solita conformazione planare.
Un numero limitato di conformazioni “proibite” di residui particolari può essere tollerato in un polipeptide se la conformazione adottata, nel suo complesso, è energeticamente favorevole. Un polipeptide tenderà a piegarsi in modo tale da adottare la conformazione più stabile. In questa conformazione, il polipeptide minimizza la sua energia libera. Nelle prossime sezioni, vedremo questo livello più alto di struttura proteica.