- Definizioni e Prevalenza
- Diagnosi
- Manifestazioni NCCAH nelle donne
- Manifestazioni nell’infanzia
- Manifestazioni nell’adolescenza e nella vita adulta
- Gestione di NCCAH Dalla prima manifestazione alla vita adulta
- Gestione durante l’infanzia
- Gestione durante l’adolescenza
- Pazienti trattati fin dall’infanzia
- Femmine con prima manifestazione durante l’adolescenza o sintomi iperandrogenici dopo l’interruzione del trattamento
- NCCAH e riproduzione
- Subfertilità
- Aborti spontanei
- Pianificazione della fertilità
- Trattamento prenatale di CAH
- Trattamento durante la gravidanza
- Altri problemi speciali
- Gestione dello stress in NCCAH
- Aspetti psicobiologici in NCCAH
- Conclusioni
- Contributi degli autori
- Dichiarazione sul conflitto di interessi
Definizioni e Prevalenza
iperplasia surrenale Congenita (CAH), comprende una famiglia di malattie autosomiche recessive caratterizzato da una lieve profondamente alterata sintesi di cortisolo a causa di un deficit in una delle cinque surrenale steroidogenica gli enzimi necessari per la produzione di cortisolo (1, 2). Convenzionalmente, CAH è diviso in (a) classica (CCAH), si presenta con sale di spreco o di semplice virilizzanti forma che si manifesta alla nascita e/o nel periodo neonatale, e (b) non classica (NCCAH), che rappresenta una forma meno grave della malattia, che manca di ambiguità genitale, non è immediatamente pericolosa per la vita, e si presenta più tardi nella vita, o rimane asintomatica, o è mal diagnosticato come malattia diverse (3). Tuttavia, i confini tra le diverse forme di CAH non sono strettamente definiti, il che tende ad aumentare le sfide associate a questo disturbo. È quindi consigliabile considerare CAH come un continuum di fenotipi, da grave a lieve o asintomatico (3).
La forma più comune di CAH è causata da deficit di 21-idrossilasi (21OHD), con conseguente ridotta o nessuna conversione di 17 idrossiprogesterone (17 OHP) a 11 deossicortisolo e di progesterone in desossicorticosterone. Il blocco della conversione degli steroidi provoca un aumento della produzione di precursori degli androgeni, sotto stimolazione CRH-ACTH, portando a iperandrogenismo biochimico, caratterizzato da elevati livelli di 17-OHP. La forma classica della malattia, si verifica in 1 su 16.000 nati vivi in tutto il mondo. Tuttavia, va detto che l’incidenza percepita della malattia è correlata al metodo di screening utilizzato. Ad esempio, è stato riportato che l’incidenza della forma classica di CAH in Svezia è di 1:11.500 quando viene utilizzato un approccio di indagine del caso, che tuttavia scende a 1:9.800 quando viene applicato lo screening ormonale. Allo stesso modo, l’incidenza in Francia e Svizzera varia tra 1:15.472 e 1:23.000 e 1:10.749 e 1:11.661 quando vengono utilizzati i diversi metodi di screening (4).
La NCCAH è molto più frequente, si verifica in circa 1 su 1.000 caucasici e più comunemente in alcuni gruppi etnici, come gli ebrei ashkenaziti (1:27), gli ispanici (1:53), gli jugoslavi (1:62) e gli italiani (1:300) (5, 6). Tuttavia, mancano dati sulla prevalenza di NCCAH basati su diversi metodi di stima (indagine caso, screening ormonale e test molecolari). NCCAH è considerato il disturbo endocrino autosomico recessivo più comune con una frequenza portante da 1:25 a 1:10. In NCCAH dovuto 21-OHD, l’attività enzimatica residua è stimata essere circa 10-70% (7-9).
Diagnosi
In confronto alla diagnosi della forma classica della malattia, che è fatta alla nascita o durante il periodo neonatale a causa di ambiguità genitale e/o sale di perdere sintomi o attraverso programmi di screening impiegato in alcuni paesi, la maggior parte dei casi di NCCAH non sono facilmente rilevabili (4, 10). Inoltre, molti individui rimangono asintomatici durante l’infanzia e l’adolescenza, hanno una normale funzione riproduttiva e diventano consapevoli del NCCAH solo a causa della diagnosi di un altro membro della famiglia e dei conseguenti test (11). Tuttavia, la maggior parte delle donne con NCCAH cercano assistenza medica quando sperimentano sintomi di eccesso di androgeni e, quando il sospetto clinico richiede test, elevati livelli basali di 17 OHP saranno più probabili che non indicano una diagnosi di NCCAH.
Infatti, le linee guida cliniche proposte dalla Endocrine Society raccomandano un valore basale non stimolato di 17 OHP come strumento di screening per NCCAH. Mattina 17 livelli di OHP > 6 nmol / L nella fase follicolare nelle femmine mestruate dovrebbero indurre un’ulteriore valutazione, poiché è stato dimostrato che livelli superiori a 6 nmol / L catturano il 90% degli individui NCCAH (12, 13). Misurazioni casuali di 17 OHP non hanno dimostrato di essere utili, poiché questi spesso producono livelli normali nei pazienti con NCCAH, e sono, inoltre, estremamente elevati nella fase luteale in metà delle femmine sane (13). Tuttavia, nei nostri dati derivati da 280 soggetti con la malattia, sei pazienti (2.1%) aveva un valore basale di 17 OHP< 6 nmol/L (14). Inoltre, Bidet et al., in una grande coorte di donne con NCCAH verificato da tecniche molecolari anche trovato basale 17 OHP valori inferiori a 6 nmol / L in 8% dei soggetti studiati (15). Infine, sulla base dei dati raccolti da Speiser et al., il 9% di individui con NCCAH ha mostrato 17 valori di OHP più bassi di 2 ng / ml (che corrisponde a 6nmol / L). Secondo altri studi, un valore basale di 17 OHP tra 5,1 e 9 nmol/L è sufficiente per la diagnosi di NCCAH (13, 16, 17). Recentemente, un livello di basale 17 OHP di 4.6 nmol / L è stato suggerito come soglia per il test ACTH per predire NCCAH in soggetti con adrenarche prematuro durante l’infanzia (18).
La somma totale di questi risultati e suggerimenti indica che la selezione dei pazienti che verranno sottoposti a un test di stimolazione Synacthen deve essere valutata caso per caso. Per la diagnosi è necessario un livello di post-stimolazione di 17 OHP superiore a 3 nmol / L (19). Gli eterozigoti portatori di una mutazione CYP21A2 presentano livelli leggermente elevati di 17 OHP dopo la stimolazione dell’ACTH, sebbene vi sia sovrapposizione in soggetti non affetti (9). Tuttavia, come Dacou-Voutetakis et al. hanno sottolineato, se la somma dei valori di OHP basale e post-stimolazione 17 supera 1,5 nmol / L, allora la possibilità di eterozigosità è eccezionalmente alta (20).
Un’altra importante considerazione riguarda le tecniche utilizzate per la valutazione di 17 OHP. 17 livelli di OHP sono misurati con una varietà di metodi di immunodosaggio, ma come è stato recentemente dimostrato, i risultati più accurati e affidabili sono stati raggiunti mediante l’implementazione della combinazione di cromatografia liquida con spettrometria di massa (LC-MS/MS). Infatti, molte misurazioni false positive 17 OHP sono state trovate quando le misurazioni LC-MS/Ms sono state confrontate con metodi standard (21) Tuttavia, queste ultime procedure non sono universalmente utilizzate, in caso di diagnosi incerta, forniranno risultati più precisi. Da notare che le soglie di screening e diagnostica per LC-MS / MS quantificate 17 OHP devono ancora essere definite. È probabile che queste soglie siano inferiori a quelle stabilite con i saggi immunologici a causa della maggiore specificità di LC-MS/MS, un metodo meno soggetto a reattività crociata e interferenze (22, 23). Resta da chiarire se sia necessario un profilo steroideo urinario per la diagnosi definitiva (22). Nei casi borderline, è consigliabile ottenere un profilo adrenocorticale completo dopo il test di stimolazione ACTH per differenziare il deficit di 21-idrossilasi da altri difetti enzimatici e stabilire una diagnosi definitiva.
In particolare, un profilo steroide completo deve essere eseguito in casi equivoci per confermare la carenza di 21-idrossilasi ed escludere altri rilevamenti enzimatici. L’inclusione di 17 OHP, cortisolo,11-deossicorticosterone,11-deossicortisolo, 17-OH-pregnenolone, deidroepiandrosterone e androstenedione in un profilo di steroidi sierici sarebbe utile per escludere altre cause di CAH come il deficit di 11β-idrossilasi e, più raramente, il deficit di 3β-idrossisteroide deidrogenasi o il deficit di ossidoreduttasi P450 (24). Mentre il test genetico non è considerato uno strumento diagnostico primario per NCCAH in questo momento, è obbligatorio per la conferma della diagnosi e per la consulenza genetica (3).
Per quanto riguarda i livelli di cortisolo, generalmente è previsto un valore post-stimolazione di almeno 496 nmol/L. Da notare, secondo Stoupa et al., il 60% di 47 bambini con NCCAH come risultato di 21 OHD ha avuto valori bassi di cortisolo dopo la stimolazione, una scoperta che indica la necessità di una maggiore sorveglianza per lo sviluppo di insufficienza surrenalica durante gli eventi di stress principali (25).
Manifestazioni NCCAH nelle donne
L’espressione clinica di NCCAH è caratterizzata da un alto livello di polimorfismo per quanto riguarda non solo l’età di esordio, ma anche i diversi segni e sintomi. È stato riferito che la prima presentazione clinica di NCCAH è nell ‘11% dei casi prima dei 10 anni e nell’ 80% tra i 10 e i 40 anni (12). La correlazione genotipo-fenotipo in CAH e NCCAH non è stata ancora chiarita. Speiser et al. suggeriscono che la maggior parte ma non tutta la variabilità fenotipica nel deficit di 21-idrossilasi deriva dalla variazione allelica nel CYP21A2 (26).
Manifestazioni nell’infanzia
Nel periodo neonatale, tipicamente le femmine NCCAH rimangono asintomatiche e hanno genitali esterni normali. Il primo caso di NCCAH riportato è una bambina di 6 mesi che ha sviluppato peli pubici (11). Di solito, i risultati clinici e i sintomi nei casi di NCCAH iniziano dall’età di 5 anni o anche più tardi e sono correlati ad un aumento dei livelli di androgeni, anche se in alcuni casi può verificarsi anche una lieve carenza di cortisolo (8). Nel 92% dei casi di NCCAH, il primo sintomo che si manifesta è il pubarca prematuro (PP), che si verifica sotto gli 8 anni di età nelle ragazze e sotto i 9 anni nei ragazzi (12) con una prevalenza stimata tra il 10 e l ‘ 11,3% (15). Da notare, nei bambini riferiti con PP, secondo diversi studi, l’incidenza di NCCAH varia dal 5 al 30% (20, 27). Tuttavia, sulla base della nostra analisi di 280 pazienti con conferma molecolare di NCCAH, abbiamo scoperto che l’incidenza di PP in 94 femmine di età inferiore agli 8 anni era pari all ‘ 88%. D’altra parte, un’analisi di 45 maschi con NCCAH ha identificato PP solo nel 29% dei soggetti (14). Inoltre, dobbiamo tenere presente che circa la metà dei soggetti con PP possono essere portatori eterozigoti di una mutazione del CYP21A2.
Un altro aspetto da chiarire è la relazione tra PP e pubertà precoce. Si ipotizza che in alcuni casi, l’aromatizzazione periferica degli androgeni surrenali agli estrogeni possa attivare l’asse ipotalamo-ipofisi-gonadico, portando alla pubertà precoce secondaria (28). In tal caso è necessario un follow-up regolare. Alcuni bambini sviluppano irsutismo, acne e / o crescita rapida e manifestano una struttura alta e un avanzamento dell’età ossea (29). Quest’ultimo può provocare un’altezza finale troncata come conseguenza della rapida fusione epifisaria. Altri rari risultati clinici durante l’infanzia includono adesione labiale, capelli perianali, clitoromegalia e raucedine della voce (30-32).
Manifestazioni nell’adolescenza e nella vita adulta
Durante l’adolescenza e l’età adulta, NCCAH nelle donne presenta sintomi iperandrogenici simili alla sindrome dell’ovaio policistico (PCOS) (33). I sintomi includono irsutismo, acne, alopecia androgenetica, anovulazione, disfunzione mestruale e infertilità. In uno studio multicentrico, i sintomi più comuni tra le donne adolescenti e adulte erano irsutismo (59%), oligomenorrea (54%) e acne (33%). Tra le 161 donne con NCCAH, i sintomi presentavano irsutismo (78%), disfunzione mestruale (54,7%) e diminuzione della fertilità (12%) (34). Nel nostro gruppo di 161 donne con la malattia il 63% del paziente ha presentato un fenotipo simile all’ovaio policistico (14). Questa scoperta ha un’implicazione viceversa, poiché l’incidenza di NCCAH è alta nelle donne con iperandrogenemia. Certamente, lo studio di Witcel et al. la segnalazione di un’incidenza di NCCAH che varia da 1 a 33% nelle donne iperandrogene è di grande interesse. In un altro studio di 950 donne che sono state indirizzate per la valutazione dell’eccesso di androgeni, NCCAH è stato rilevato nel 4,2% di esse. Tuttavia, i pazienti NCCAH erano più giovani e più irsuti rispetto agli altri gruppi di pazienti iperandrogeni. Erano inoltre caratterizzati da livelli significativamente più elevati di testosterone, testosterone libero e 17 OHP rispetto ai pazienti con altre sindromi iperandrogeniche. Nell’ecografia ovarica, il 77% di essi ha mostrato ovaie policistiche e il 41% ha aumentato le dimensioni ovariche. In sintesi, hanno soddisfatto i criteri PCOS secondo NIH e Rotterdam con una percentuale rispettivamente del 56 e del 72,8% (35).
La natura progressiva della malattia è evidenziata dal fatto che la prevalenza dell’irsutismo ha dimostrato di aumentare con l’età ed è stata osservata essere rara prima della pubertà. L’irsutismo è la manifestazione clinica più comune riportata nei pazienti con NCCAH, che varia dal 71 al 96% (28, 36). Le ragazze puberali con NCCAH sono tipicamente presenti con irsutismo (37, 38). All’altra estremità dello spettro c’è il problema dell’alopecia. Nei pazienti con NCCAH è stata riportata calvizie maschile. Anche la prevalenza dell’alopecia sembrava aumentare con l’età, dal 6% nei pazienti durante la seconda decade di vita al 19% nella quinta, indicando nuovamente la natura progressiva della malattia (39). L’acne è riportata in quasi il 33% dei soggetti NCCAH (36). Sorprendentemente, mutazioni del gene CYP21A2 che causa NCCAH sono state rilevate nel 4,9% di 123 femmine adulte che presentavano acne grave (40).
Irregolarità mestruali tra cui oligomenorrea o amenorrea secondaria possono spesso essere il segno di presentazione di NCCAH in individui post menarca. Inoltre, l ‘ 8-9% dei casi sperimenta anche l’amenorrea primaria e, per questo motivo, consultare un medico per la prima volta. Inoltre, tra gli individui NCCAH, l’oligomenorrea è più comune dell’amenorrea primaria. Ad esempio, in una serie di Moran et al., tra le donne NCCAH di età compresa tra 10 e 19 anni, l’oligomenorrea era presente nel 56% dei casi rispetto al solo 9% dell’adolescenza con amenorrea primaria (12). Per concludere, così tanti sintomi di NCCAH assomigliano a quelli di PCOS che non sorprende che NCCAH sia stato soprannominato il grande mimicker di PCOS. In ogni caso, una diagnosi di NCCAH deve essere presa in considerazione durante la valutazione di qualsiasi giovane donna che viene indirizzata per sintomi iperandrogenici.
In uno studio di Arlt et al., i pazienti con NCCAH sono stati caratterizzati da BMI significativamente più alto e altezza finale più bassa in confronto a controlli abbinati (41). Questi dati, in combinazione con alcuni studi che indicano la presenza di insensibilità all’insulina, suggeriscono un profilo metabolico sfavorevole che potrebbe essere spiegato patofisiologicamente dall’effetto di elevate concentrazioni di androgeni e/o come risultato della terapia con glucocorticoidi (42, 43). L’osteoporosi può anche essere rilevata in questi soggetti, probabilmente come conseguenza della terapia con corticosteroidi (34).
Gestione di NCCAH Dalla prima manifestazione alla vita adulta
Una volta stabilita la diagnosi di NCCAH, diverse questioni relative alle successive raccomandazioni terapeutiche meritano considerazione. La prima domanda da affrontare è se sia indicata la terapia con glucocorticoidi (GC). Il ragionamento generale alla base di questo approccio è che fornendo concentrazioni sufficienti di cortisolo al paziente, i suoi bisogni quotidiani sono coperti e, di conseguenza, la stimolazione dell’asse CRH-ACTH sarà rastremata, portando a una diminuzione della produzione di androgeni surrenali. La regola generale relativa a questo approccio è che le GCS vengono somministrate a dosi sostitutive e non farmacologiche, mentre l’influenza di età, sesso, dati di laboratorio, raccomandazioni specifiche per il paziente e obiettivi del trattamento sulla terapia sostitutiva con glucocorticoidi sono seriamente presi in considerazione. Tuttavia, dobbiamo anche essere consapevoli che la terapia sostitutiva GC prolungata può portare a ipercortisolemia, con i conseguenti effetti avversi ben documentati su ogni aspetto del metabolismo, in particolare la crescita, la distribuzione del grasso e la resistenza all’insulina, nonché sul profilo psicologico. Un altro svantaggio principale di questo approccio è la mancanza di un adeguato indice clinico o marcatore biochimico di un adeguato dosaggio sostitutivo, come esiste per quanto riguarda i valori di TSH nell’ipotiroidismo.
Infine, il fatto che non vi sia consenso su quale GC debba essere usato e l’assenza di dati a lungo termine riguardanti diverse modalità di somministrazione di GC complicano ulteriormente le decisioni dei medici curanti e la selezione della scelta ottimale per ciascun paziente. L’idrocortisone è usato tipicamente in bambini, poichè più strettamente assomiglia all’ormone naturale (cortisolo), ma non è considerato un approccio adatto in adolescenti ed in giovani femmine dovuto la necessità di dosaggio quotidiano multiplo. Quindi, la maggior parte degli endocrinologi adulti preferisce desametasone o prednisolone, a dosi appropriate. D’altra parte, va sottolineato che l’equivalenza di diversi GCS si basa sulla loro azione antinfiammatoria e non su diversi aspetti del metabolismo umano. Poiché il cortisolo colpisce quasi il 20% del genoma umano, sono previste diverse risposte di diversi GCS in vari tessuti. A corroborare questa percezione è il fatto che la somministrazione di desametasone a pazienti con CAH ha mostrato un deterioramento degli indici di insulino-resistenza rispetto ad altri GCs (44). Inoltre, la somministrazione di 2,5-7,5 mg di prednisolone, una dose considerata normale, esercita un impatto negativo di lunga data sul metabolismo osseo (45). Pertanto, l’idrocortisone deve essere considerato la migliore forma di trattamento nei casi di terapia di integrazione GC. L’avvento della formula di idrocortisone di nuova sintesi con una pillola al giorno e i suoi risultati positivi iniziali nei pazienti con CAH mostrano molte promesse per il futuro (46).
Gestione durante l’infanzia
NCCAH I pazienti che vengono diagnosticati durante l’infanzia con segni di PP possono essere trattati con idrocortisone con lo scopo di sopprimere gli ormoni surrenali e prevenire il rapido avanzamento dell’età ossea che potrebbe influenzare l’altezza finale. In quei pazienti in cui il trattamento con idrocortisone è stato iniziato 1 anno prima dell’inizio della pubertà e che avevano un’età ossea inferiore a 9 anni, l’altezza finale è rimasta entro il potenziale genetico (47). Gli studi disponibili indicano che l’altezza dell’adulto si è avvicinata all’altezza target prevista in pazienti che sono stati strettamente monitorati e che hanno rispettato rigorosamente i piani farmacologici (48-50). Un recente piccolo studio su cinque casi (pazienti di età compresa tra 6,1 e 9,2 anni) ha dimostrato che una dose ultrabassa di desametasone è un’opzione promettente per ridurre lo stress endogeno e i suoi effetti. Resta da chiarire se questo costituisca un approccio realistico e quanto debba essere la dose somministrata (51). Uno studio di Nebesio et al. ha confrontato gli effetti ormonali e la farmacocinetica di idrocortisone, prednisolone e desametasone in 9 pazienti prepuberali con CCAH. Hanno dimostrato che il desametasone era più potente delle altre forme nel raggiungere livelli ormonali surrenali significativamente più bassi, suggerendo quindi che il desametasone è più efficace per la soppressione della produzione di androgeni surrenali (46). Naturalmente, sono necessari ulteriori studi per verificare o rifiutare questa scoperta. Inoltre, in alcuni casi, concentrazioni elevate di androgeni possono portare alla stimolazione secondaria dell’asse GnRH, portando a pubertà prematura. In tal caso, può essere prescritto un percorso parallelo con l’analogo GnRH se l’età ossea è significativamente superiore all’età cronologica e/o l’altezza finale proiettata è sproporzionata rispetto all’altezza target.
Un’altra importante considerazione riguarda i casi di concomitante carenza di GH nel caso in cui il trattamento con analoghi del GnRH e la prescrizione di GH abbiano portato al raggiungimento dell’altezza target (52). Secondo le attuali linee guida, la terapia sopra descritta è raccomandata con cautela solo per quei casi in cui l’altezza prevista SD è -2.25 < l’altezza target o anche inferiore (53). Un’indicazione alternativa per iniziare il trattamento con idrocortisone è una risposta inadeguata al cortisolo dopo la stimolazione ACTH (36).
Il trattamento GC preferito nei bambini è di solito idrocortisone 10-15 mg/m2, diviso in tre dosi. Spesso, anche dosi più basse si sono dimostrate efficaci, a partire da 6 mg / m2 / die (34). Il sovradosaggio deve essere evitato, considerando che può causare scarsa crescita e caratteristiche Cushingoidi. Il modello di crescita regolare, un’età ossea compatibile con l’età cronologica e l’assenza di obesità centrale possono anche servire come indici clinici per una gestione appropriata. Per quanto riguarda il profilo biochimico/ ormonale, i livelli di androstenedione e testosterone nelle gamme medio-superiori per l’età ossea sono considerati migliori marcatori di un’adeguata terapia sostitutiva GC nei bambini con NCCAH. Tuttavia, i livelli di testosterone soppressi sono stati trovati nel 10% dei pazienti NCCAH, mentre un altro 28% dei pazienti aveva aumentato le concentrazioni di testosterone, questo fenomeno probabilmente attribuibile alla variabilità dell’idrocortisone (54, 55). I valori di DHEAS sono diminuiti con dosi molto piccole, mentre la soppressione di 17 livelli di OHP e progesterone richiede dosi molto elevate di GC. Va notato che 17 valori di OHP sono cruciali per la diagnosi, ma non sono utili durante il follow–up. Da notare, il prelievo di sangue per la valutazione ormonale deve essere effettuato senza interruzione della terapia. Tuttavia, a causa della perplessità della malattia e della sua natura multiforme, non ci sono linee guida specifiche per i tempi dei cambiamenti del regime o la cessazione della terapia con glucocorticoidi nei bambini.
Gestione durante l’adolescenza
Pazienti trattati fin dall’infanzia
Fino alla creazione del normale schema mestruale nelle ragazze NCCAH, la continuazione di GCs iniziata durante l’infanzia è altamente raccomandata (56). Tuttavia, i pazienti adolescenti spesso non mostrano sufficiente conformità con la somministrazione cronica di farmaci e spesso omettono le dosi. Inoltre, durante la pubertà, l’emivita dell’idrocortisone diminuisce del 50% a causa dell’aumento dei livelli di IGF-1, che diminuisce l’attività di 11ßOHSD, nonché a causa dell’aumentata clearance del cortisolo derivante dall’amplificazione della velocità di filtrazione glomerulare (57). Nella giovane femmina, 2 anni dopo il menarca e se sono stati registrati normali cicli ovulatori, è altamente raccomandato un approccio centrato sul paziente verso i sintomi iperandrogenici che possono apparire.
Se ci sono gravi risultati iperandrogenici, come irsutismo, acne e/o oligomenorrea, sarà presa in considerazione la continuazione del trattamento. È importante ricordare la sensibilità fragile di un giovane adolescente, che sarà seriamente danneggiato dai segni “repellenti” dell’iperandrogenismo. La combinazione di segni iperandrogeni, disturbi mestruali e scarsa qualità della vita è ben documentata ed è particolarmente elevata nelle donne più giovani. Un altro punto da considerare è la risposta della stimolazione post ACTH della riserva surrenale. Dato che il cortisolo di picco delle femmine puberali e adulte dopo la stimolazione è inferiore a 496 nmol / L, in caso di stress, deve essere somministrato un trattamento steroideo (58). D’altra parte, se nessuno dei suddetti sintomi si verifica nel paziente giovane, il trattamento con GC può essere interrotto, dopo di che si consiglia un follow-up regolare.
Femmine con prima manifestazione durante l’adolescenza o sintomi iperandrogenici dopo l’interruzione del trattamento
In presenza di sintomi iperandrogenici, è altamente raccomandato un approccio orientato al paziente, concentrandosi sui principali disturbi del paziente. Questo è molto meglio di un modus operandi universale generale, poiché, con quest’ultimo approccio, il giovane paziente sarà deluso, questo porta alla sospensione del trattamento e, di conseguenza, a risultati negativi per la salute in età adulta. Nei casi con irsutismo severo, un corso di 6-12 mesi con le pillole contraccettive orali (OCPs) costituisce una procedura ragionevole dati gli effetti benefici di OCPs sulla produzione del fegato di SHBG e sulla diminuzione del rilascio dell’androgeno dall’ovaio. Il miglioramento clinico sarà preveduto dopo almeno 2-3 mesi dell’inizio di OCPs, mentre l’uso simultaneo di un progestagen con le proprietà androgene minime altamente è raccomandato. Se l’OCPS fallisce come approccio di prima linea, gli antiandrogeni (spironolattone, flutamide, bicalutamide, ciproterone acetato e finasteride) possono essere aggiunti al trattamento. Nella nostra esperienza, la somministrazione di bicalutamide ha ottenuto un miglioramento significativo nei casi di acne grave, ma risultati simili non sono stati ottenuti nei casi con grave irsutismo. Approcci cosmetici come l’applicazione laser e i depilatori possono anche essere suggeriti per le donne che lamentano una crescita eccessiva o indesiderata dei capelli o una perdita di capelli del cuoio capelluto (alopecia androgenetica) (32, 34, 57).
In pazienti con insufficiente secrezione di cortisolo dopo stimolazione, o se OCPs e antiandrogeni non possono essere tollerati, un ciclo con GCs è altamente raccomandato (57). Dati da New et al. indicare che le mestruazioni irregolari e l’acne sono invertite entro 3 mesi dall’inizio della terapia con glucocorticoidi, mentre l’irsutismo richiede quasi 30 mesi (59). A differenza dell’infanzia, nell’adolescenza vengono spesso utilizzati steroidi ad azione prolungata e si raccomandano regimi di 5 mg di prednisolone o 0,25 mg di desametasone (53). Tuttavia, nel mondo reale i dati clinici hanno mostrato una varietà di diversi regimi applicati nella gestione NCCAH (41).
L’obiettivo primario del trattamento deve essere il sollievo del paziente dai sintomi iperandrogenici. Pertanto, tra gli individui con diagnosi NCCAH basata esclusivamente sui risultati di laboratorio, le caratteristiche cliniche dovrebbero guidare le raccomandazioni di gestione, poiché i risultati ormonali e molecolari non prevedono necessariamente la gravità clinica. Secondo le linee guida della Endocrine Society, ai pazienti con NCCAH deve essere data la possibilità di interrompere la terapia con GC quando i sintomi si risolvono (60). Naturalmente, questi pazienti non devono essere persi per il follow-up, mentre il trattamento deve essere ripreso in caso di recidiva dei sintomi. Inoltre, in caso di interruzione del trattamento, i pazienti devono essere informati sulla possibilità di infertilità e devono essere incoraggiati a consultare un medico se desiderano concepire (19). Da notare, il trasferimento appropriato del paziente dall’endocrinologo pediatrico all’adulto deve essere effettuato, in modo ottimale dopo 1 anno di monitoraggio sincronizzato (53, 61).
NCCAH e riproduzione
Subfertilità
La subfertilità è comunemente riportata nelle forme classiche di CAH, che è principalmente dovuta a disturbi mestruali, anovulazione cronica e deformità anatomiche. Il tasso di natalità è stato stimato al 17% rispetto al 65% della popolazione di controllo (62). Nel frattempo, i dati relativi alla fertilità nelle donne con NCCAH sono stati recentemente valutati in dettaglio e l’incidenza di infertilità stimata è dell ‘ 11% tra le donne NCCAH, cioè relativamente più mite rispetto a CAH, e in molti casi è facilmente risolvibile. Bidet et al. valutata la fertilità in 190 donne affette da NCCAH, 95 delle quali volevano rimanere incinta. In questa popolazione, sono state segnalate 187 gravidanze in 85 donne, che hanno portato a 141 nascite in 82 individui. Va evidenziato che 99 delle gravidanze (52,9%) si sono verificate prima della diagnosi di NCCAH, tre delle quali con l’applicazione di protocolli di induzione dell’ovulazione, il resto è spontaneo, mentre 88 gravidanze hanno avuto luogo dopo la diagnosi di NCCAH. Nella stragrande maggioranza di loro la gravidanza si è sviluppata dopo l’istituzione della terapia con idrocortisone, mentre in 11 donne è avvenuta spontaneamente (63, 64). In un altro rapporto di 22 donne NCCAH osservate che desideravano una gravidanza, 12 gravidanze sono seguite con prednisone (65).
Aborti spontanei
Nella coorte di Bidet et al., il tasso di aborto spontaneo era 6.5 e 26.3% in pazienti trattati con GCs e pazienti non trattati, rispettivamente (64). L’esito della gravidanza può anche avere successo senza alcun trattamento con glucocorticoidi nei casi in cui NCCAH non è stato ancora diagnosticato, come riportato anche in un case report di Cuhaci et al. Tuttavia, la stessa coppia ha sperimentato due aborti spontanei e ha riportato subfertilità dopo questa prima gravidanza (66). Per quanto riguarda la possibilità di gravidanze pretermine, come valutato da Bidet et al., non differisce significativamente da quello del resto della popolazione (15).
Pianificazione della fertilità
Per quelle donne con iperandrogenismo sintomatico o con infertilità segnalata ma che desiderano concepire, la terapia con GC è altamente raccomandata. Durante questo periodo, i glucocorticoidi utilizzati sono prednisone o idrocortisone. L’azione più importante, perché guiderà i passaggi successivi, è il test genetico dei futuri genitori. Le femmine con NCCAH che desiderano concepire dovrebbero essere consapevoli del rischio di dare alla luce un bambino con la forma classica della malattia. L’esito delle gravidanze tra le donne con NCCAH, e più specificamente l’incidenza di bambini nati con CCAH, è stimato in studi recenti tra l ‘ 1,5 e il 2,5%, con NCCAH a circa il 15% (36, 64). Naturalmente, questa frequenza dipende dalla popolazione di riferimento, una maggiore incidenza che si verifica in popolazioni con alti tassi di matrimoni misti (36). Le probabilità previste di genitori che danno alla luce un bambino con CAH sono 1 su 120 per il genotipo paterno sconosciuto, zero se il padre non ha alcuna mutazione rilevante e 1 su 4 se ha una mutazione eterozigote (19). Di conseguenza, il test genetico dei partner di queste donne è essenziale per valutare il rischio di dare alla luce un bambino con la forma classica di CAH (64, 67).
Nel caso in cui la futura madre porti due mutazioni NC (Tabella 1), non vi è alcuna necessità assoluta di test genetici nel futuro padre, poiché, se è eterozigote per le mutazioni del CYP12A2, la prole è a rischio di sviluppare la forma non classica della malattia in futuro. Tuttavia, va notato che lo stato del vettore è del 2-15% in diverse popolazioni e la metà di questi individui porta una grave mutazione. Inoltre, le raccomandazioni definitive relative alle situazioni in cui non è richiesto il test genetico sono difficili data la correlazione genotipo-fenotipo imperfetta, in particolare per le mutazioni più lievi . Ad esempio, la mutazione P30L, più frequentemente associata a NCCAH, si verifica anche in pazienti con CAH classica . Un ampio studio europeo multicentrico ha recentemente dimostrato che questo era il caso nel 78% dei pazienti . In questo studio, la lieve mutazione V218L è stata associata al Classico piuttosto che al NCCAH nel 30% dei casi. Quindi tutti i pazienti con NCCAH dovrebbero essere offerti consulenza genetica e valutazione molecolare dei partner riproduttivi.
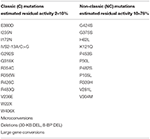
Tabella 1. Mutazioni di CYP21A2 con attività enzimatica residua minima (C) o moderata (NC) (14, 68, 69).
Al contrario, nel caso in cui la futura madre sia un eterozigote composto con una grave (C) e una mutazione NC, è obbligatorio il test genetico del futuro padre. Dobbiamo tenere presente che i dati provenienti da studi che utilizzano immunodosaggi o le procedure LC-MS/MS più accurate, evidenziano costantemente l’incapacità di escludere in modo affidabile l’eterozigosità utilizzando valori di OHP 17 stimolati basali o ACTH, a causa della significativa sovrapposizione con l’intervallo normale. Si può suggerire l’uso di un test Synacthen, e se non è compatibile con l’eterozigosità (somma di valori di 17 OHP stimolati basali e di picco < 1,5 nmol/L), il test del DNA potrebbe essere evitato. Tuttavia, dovremmo essere molto cauti nell’interpretazione di questo test, poiché non è stato convalidato in altri studi. Quindi, l’uso di ACTH stimolato 21-deoxycortisol singolarmente o come parte di un rapporto steroide o profilo steroide, può facilitare l’identificazione biochimica degli eterozigoti in futuro, in particolare come LC-MS / MS diventa più ampiamente disponibile . Se il futuro padre porta una mutazione NC, allora non è necessario nient’altro. Viceversa, se è portatore di una mutazione classica, sorgerà la questione relativa al trattamento prenatale del feto. Tutte le possibili combinazioni di genotipo e le procedure suggerite sono presentate nella Tabella 2.

Tabella 2. Pianificazione delle procedure suggerite durante la gravidanza in base al genotipo dei futuri genitori.
Trattamento prenatale di CAH
Lo scopo del trattamento prenatale di CAH è la prevenzione della virilizzazione genitale del feto, ma anche l’alleviamento dello stress avvertito dai genitori che rischiano di avere un figlio con genitali ambigui (70). Il desametasone è usato a causa della sua capacità di attraversare la placenta e perché non è disattivato dalla deidrogenasi steroide placentare 11β OH e si lega solo minimamente alla globulina legante il cortisolo del sangue della madre (CBG). Desametasone, sopprimendo la secrezione fetale di ACTH, diminuisce la produzione fetale elevata dell’androgeno. Il trattamento deve essere interrotto nel caso in cui l’analisi del cariotipo o del DNA riveli rispettivamente un maschio o una femmina non affetta (71). Tuttavia, sebbene la virilizzazione genitale fetale sia già iniziata a 6-7 settimane dopo il concepimento, le biopsie villose corioniche per la diagnosi genetica non possono essere ottenute prima della 10a−12a settimana. Questo intervallo di tempo suggerisce che tutte le gravidanze a rischio di virilizzare CAH dovrebbero essere trattate, anche se solo 1 su 8 feti è affetto e femmina (19, 34, 70).
Rimane controverso se questa esposizione del feto al desametasone per misure preventive sia accettabile dal punto di vista medico ed etico (70). Studi su animali hanno dimostrato che il desametasone del primo trimestre diminuisce il peso alla nascita, influenza la funzione delle cellule beta pancreatiche renali e lo sviluppo del cervello e predispone ad ansia, ipertensione e iperglicemia in età adulta (70). Inoltre, il 10-20% delle donne in gravidanza che utilizzano il trattamento con desametasone durante la gravidanza si lamentano di aumento di peso, aumento dell’appetito, sbalzi d’umore, insonnia, edemi, ipertensione, iperglicemia e strie. Tuttavia, New et al. trovato un punteggio di Prader inferiore alla media nei feti trattati con desametasone prenatale, ma nessuna differenza nei risultati a lungo termine (72). Inoltre, in una recensione di Merce Fernandez-Balsells et al., il desametasone è stato indicato per essere associato con riduzione nella virilizzazione del feto senza gli effetti contrari materni o fetali significativi (63). Tuttavia, gli autori della suddetta revisione e le linee guida consecutive della Endocrine Society sottolineano che a causa delle piccole dimensioni del campione in tutto il corpo della letteratura, l’argomento rimane incerto e sono chiaramente necessarie ulteriori indagini.
La decisione di iniziare il trattamento deve essere intrapresa solo in grandi centri con un team esperto e protocolli approvati da commissioni di revisione istituzionale e basati sui valori e le preferenze della famiglia e con il loro consenso informato scritto come prerequisito (19, 63). Diversi specialisti nel campo indicano che un valore più alto dovrebbe essere posto sulla prevenzione dell’esposizione prenatale non necessaria della madre e del feto al desametasone piuttosto che imporre il pedaggio emotivo dei genitali ambigui su genitori e pazienti. Soprattutto, dovrebbe essere chiarito ai genitori che la somministrazione di desametasone non modifica lo stato del paziente, ma è diretta a ridurre la necessità di un intervento chirurgico piuttosto che preservare la vita o la capacità intellettuale. Devono anche sapere che la probabilità che il loro bambino soffra della forma classica della malattia è alta, nonostante il trattamento. Soprattutto, nel frattempo, l’esposizione di un giovane organismo a un GC molto potente durante un periodo particolarmente delicato di programmazione e crescita fetale, che potrebbe rivelarsi inutile nel caso di un feto con il cariotipo XY, non è attualmente supportata da dati robusti e indiscutibili.
Idealmente, dovrebbe esserci una diagnosi precoce, eseguita tramite una tecnica non invasiva e prima dell’inizio della virilizzazione genitale del feto. Lavorando in questa direzione, nuovi studi indicano l’uso del DNA fetale senza cellule ottenuto dal plasma materno come metodo promettente che consentirà la determinazione del sesso fetale e la diagnosi di CAH in età gestazionale precoce (<9 settimane). Se questa procedura è ampiamente implementata nella pratica clinica, si eviterà un trattamento prenatale con desametasone non necessario (73).
Trattamento durante la gravidanza
Durante la gravidanza, le donne con NCCAH devono essere trattate preferibilmente con idrocortisone, non solo perché la gravidanza è tipicamente accompagnata da elevati livelli di stress, ma anche come misura preventiva contro l’elevata incidenza di aborti spontanei sopra menzionata. Questa forma di glucocorticoide è favorita a causa della sua metabolizzazione dall’enzima placentare 11β OH steroide deidrogenasi II, che impedisce al glucocorticoide di avere alcun effetto sul feto (71). Al momento del concepimento, la dose di idrocortisone deve essere aumentata a 20-25 mg / die e le modifiche della dose vengono effettuate ogni 6-8 settimane con l’obiettivo di mantenere i livelli di testosterone ai livelli normali superiori del trimestre di gravidanza (74). Ad esempio, vengono mostrati i valori di testosterone e D4 di un paziente con NCCAH dal concepimento fino a 3 anni dopo il parto di due gemelli sani (Figura 1).
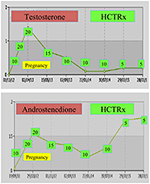
Figura 1. Livelli di testosterone e andostenedione e successivo dosaggio di idrocortisone (HCTRx), dal concepimento fino a 3 anni dopo il parto somministrato a un paziente con NCCAH.
Altri problemi speciali
Gestione dello stress in NCCAH
Durante lo stress grave, un intervento chirurgico o una malattia grave, i pazienti con NCCAH trattati con glucocorticoidi possono richiedere dosi maggiori o più frequenti di glucocorticoidi dato che la loro funzione surrenale è soppressa iatrogenicamente. È quindi fondamentale educare i genitori dei bambini piccoli, nonché rieducare i pazienti sulla loro transizione verso l’assistenza agli adulti, sul dosaggio dello stress. Per i pazienti con NCCAH che non sono trattati con GCs o in caso di interruzione del trattamento, deve essere valutata la risposta del cortisolo all’ACTH. Quasi un terzo dei pazienti NCCAH risponde in modo inadeguato alla stimolazione (15, 75). Per coloro che rispondono normalmente alla stimolazione ACTH non è raccomandato alcun trattamento con dosi di stress (19). La terapia con mineralcorticoidi non è richiesta in nessuno dei casi, poiché questi pazienti hanno una normale secrezione di aldosterone e non sviluppano spreco di sale (58). Da notare, nel caso di ipertiroidismo o di un aumento del trattamento con levotiroxina, la clearance del cortisolo aumenta e può verificarsi una crisi surrenalica (58, 76).
Aspetti psicobiologici in NCCAH
Meyer-Bahlburg et al. hanno riportato in diversi studi una compromissione del profilo psicologico tra i pazienti con NCCAH a causa della carenza di 21OH. Più specificamente, le donne colpite avevano un punteggio di mascolinizzazione/defeminizzazione più elevato su diverse misure di comportamento correlato al genere rispetto alle normali donne di controllo, anche se nettamente inferiore rispetto alle donne con CAH classica. Avevano anche aumentato significativamente l’orientamento bisessuale o omosessuale. Interviste clinico-qualitative retrospettive con queste donne hanno rivelato una storia di disagio e stress sociale legati alle loro esperienze di pre-trattamento con segni androgeni-dipendenti, come acne, irsutismo e difficoltà di concepimento (59, 77, 78). Allo stesso modo, Arlt et al. ha mostrato che lo stato di salute soggettivo era significativamente compromesso in tutti gli otto domini di un sondaggio sulla salute a breve termine, con le differenze più importanti, rispetto ai controlli abbinati all’età e al sesso, relativi ai domini salute generale, vitalità e limitazioni di ruolo a causa di problemi emotivi. Il questionario Hospital Anxiety and Depression Scale (HADS) ha anche rivelato un aumento dei punteggi di ansia (41). Tenendo conto di questi risultati, i parametri psicologici per guidare la terapia devono essere considerati nelle donne con NCCAH e, nel contesto dell’approccio orientato al paziente, è necessario offrire una diagnosi psicologica e un supporto.
Conclusioni
NCCAH è considerato come la forma più frequente e più mite di CAH a causa della ritenzione del 20-50% di attività enzimatica. La maggior parte dei pazienti può consultare un medico in qualsiasi fase della loro vita a causa di sintomi correlati all’eccesso di androgeni. Questi includono pubarca precoce, rapida crescita, irsutismo, acne, irregolarità mestruali o infertilità, così come molte altre manifestazioni meno prominenti. Al mattino presto si raccomandano valori basali di 17 OHP come un buon test di screening iniziale e un’ulteriore valutazione con stimolazione ACTH e, nel caso di risultati borderline, test genetici. Come regola generale, androstenedione e non 17 livelli di OHP devono essere utilizzati durante il follow-up.
La decisione di trattamento deve essere basata sulla valutazione dei fatti e deve seguire un approccio individualizzato. Il trattamento non è sempre indicato a meno che il paziente non sia sintomatico, ad esempio, bambini con esordio precoce e rapida progressione dei peli pubici e corporei, rapida crescita e/o avanzamento scheletrico, o donne con oligomenorrea, acne, irsutismo, infertilità o una combinazione di questi e altri sintomi sopra menzionati. La consulenza genetica è fortemente consigliata nelle donne NCCAH che desiderano concepire, così come la genotipizzazione del padre. La gestione complessiva del paziente include inoltre la gestione delle probabili complicanze della terapia con glucocorticoidi o delle manifestazioni correlate al metabolismo della malattia. Infine, dato che molte questioni terapeutiche relative alla gestione appropriata di questi pazienti non sono ancora state chiarite, è molto importante che il medico curante si tenga aggiornato su tutti gli sviluppi in questo campo e integri i nuovi dati nella sua pratica clinica. Certamente, ulteriori studi clinici in questo settore sono essenziali.
Per riassumere, per la donna NCCAH, l’approccio ideale è su misura, incorporando una transizione graduale della sua gestione una volta che viene indirizzata dall’endocrinologo pediatrico a quello adulto, insieme a un trattamento orientato ai sintomi che la accompagnerà per tutta la vita. Dati i molteplici fattori che influenzano il sistema iperandrogenico, è consigliabile incoraggiare i pazienti ad adottare uno stile di vita più sano migliorando le loro abitudini alimentari, aumentando l’esercizio fisico e puntando alla riduzione del peso. Inoltre, in alcuni casi, il supporto psicologico è spesso utile. Inoltre, gli specialisti nei campi coinvolti nel trattamento di questo disturbo, come dermatologi, ginecologi e psicologi, hanno bisogno di una comprensione approfondita della gestione di casi sospetti o già diagnosticati di NCCAH. Per concludere, teniamo a mente la penetrante citazione dell’astronoma americana Vera Cooper Rubin: “La scienza progredisce meglio quando le osservazioni ci costringono a modificare i nostri preconcetti.”
Contributi degli autori
Tutti gli autori elencati hanno dato un contributo sostanziale, diretto e intellettuale al lavoro, e lo hanno approvato per la pubblicazione.
Dichiarazione sul conflitto di interessi
Gli autori dichiarano che la ricerca è stata condotta in assenza di relazioni commerciali o finanziarie che potrebbero essere interpretate come un potenziale conflitto di interessi.
1. Speiser PW, PC bianco. Iperplasia surrenalica congenita. N Ingl J Med. (2003) 349:776–88. doi: 10.1056 / NEJMra021561
PubMed Abstract / CrossRef Full Text / Google Scholar
2. Turcu AF, Auchus RJ. Adrenal steroidogenesis and congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. (2015) 44:275–96. doi: 10.1016/j.ecl.2015.02.002
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Speiser PW. Nonclassic adrenal hyperplasia. Rev Endocr Metab Disord. (2009) 10:77–82. doi: 10.1007/s11154-008-9097-x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Gidlöf S, Falhammar H, Thilén A, von Döbeln U, Ritzén M, Wedell A, et al. Cento anni di iperplasia surrenalica congenita in Svezia: uno studio retrospettivo di coorte basato sulla popolazione. Lancet Diabete Endocrinol. (2013) 1:35–42. doi: 10.1016/S2213-8587(13)70007-X
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, Nuovo MI. Alta frequenza di carenza di steroidi 21-idrossilasi non classica. Sono J Hum Genet. (1985) 37:650–67.
PubMed Abstract | Google Scholar
6. Wilson RC, Nimkarn S, Dumic M, Obeid J, Azar MR, Azar M, et al. Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Mol Genet Metab. (2007) 90:414–21. doi: 10.1016/j.ymgme.2006.12.005
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Miller WL. Clinical review 54: genetics, diagnosis, and management of 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1994) 78:241–6. doi: 10.1210/jcem.78.2.8106606
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Witchel SF, Azziz R. Nonclassic congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2010) 2010:625105. doi: 10.1186/1687-9856-2010-625105
PubMed Abstract | CrossRef Full Text | Google Scholar
9. La nostra azienda si occupa di Carenza di steroidi 21-idrossilasi: il genotipo non può predire il fenotipo. J Clin Endocrinol Metab. (1995) 80:2322–9. doi: 10.1210 / cc.80.8.2322
CrossRef Full Text/Google Scholar
10. PC bianco. Screening neonatale per iperplasia surrenalica congenita. Nat Rev Endocrinol. (2009) 5:490–8. doi: 10.1038 / nrendo.2009.148
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Kohn B, Levine LS, Pollack MS, Pang S, Lorenzen F, Levy D, et al. Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. (1982) 55:817–27. doi: 10.1210/jcem-55-5-817
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Moran C, Azziz R, Carmina E, Dewailly D, Fruzzetti F, Ibañez L, et al. 21-Idrossilasi-carente iperplasia surrenalica non classica è un disturbo progressivo: uno studio multicentrico. Am J Ostetrico Gynecol. (2000) 183:1468–74. doi: 10.1067 / mob.2000.108020
PubMed Abstract | CrossRef Full Text/Google Scholar
13. Azziz R, Hincapie LA, Knochenhauer ES, Dewailly D, Fox L, Boots LR. Screening per iperplasia surrenalica non classica con deficit di 21-idrossilasi tra donne iperandrogene: uno studio prospettico. Fertil Steril. (1999) 72:915–25. doi: 10.1016/S0015-0282(99)00383-0
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Livadas S, Dracopoulou M, Dastamani A, Sertedaki A, Maniati-Christidi M, Magiakou A-M, et al. Lo spettro dei risultati clinici, ormonali e molecolari in 280 individui con iperplasia surrenalica congenita non classica causata da mutazioni del gene CYP21A2. Clin Endocrinol (Oxf). (2015) 82:543–9. doi: 10.1111 / cen.12543
PubMed Abstract | CrossRef Full Text/Google Scholar
15. Bidet M, Bellanné-Chantelot C, Galand-Portier M-B, Tardy V, Billaud L, Laborde K, et al. Caratterizzazione clinica e molecolare di una coorte di 161 donne non correlate con iperplasia surrenalica congenita non classica dovuta a deficit di 21-idrossilasi e 330 membri della famiglia. J Clin Endocrinol Metab. (2009) 94:1570–8. doi: 10.1210 / cc.2008-1582
PubMed Abstract | CrossRef Full Text/Google Scholar
16. Escobar-Morreale HF, Sanchón R, San Millán JL. Uno studio prospettico della prevalenza di iperplasia surrenalica congenita non classica tra le donne che presentano sintomi e segni iperandrogeni. J Clin Endocrinol Metab. (2008) 93:527–33. doi: 10.1210 / cc.2007-2053
PubMed Abstract | CrossRef Full Text/Google Scholar
17. Dewailly D, Vantyghem-Haudiquet MC, Sainsard C, Buvat J, Cappoen JP, Ardaens K, et al. Fenotipi clinici e biologici in carenza tardiva di 21-idrossilasi. J Clin Endocrinol Metab. (1986) 63:418–23. doi: 10.1210 / jcem-63-2-418
PubMed Abstract | CrossRef Full Text/Google Scholar
18. Gönç EN, Ozön ZA, Alikaşifoglu A, Engiz O, Bulum B, Kandemir N. (2011). Il siero basale 17-OH progesterone è un parametro affidabile per prevedere l’iperplasia surrenalica congenita non classica nell’adrenarche prematuro? Turk J Pediatr. 53:274–80.
PubMed Abstract / Google Scholar
19. Il suo nome deriva dal greco antico, che significa “la parola di Dio”. Iperplasia surrenalica congenita dovuta a carenza di steroidi 21-idrossilasi: una linea guida sulla pratica clinica della società endocrina. J Clin Endocrinol Metab. (2010) 95:4133–60. doi: 10.1210 / cc.2009-2631
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Dacou-Voutetakis C, Dracopoulou M. High incidence of molecular defects of the CYP21 gene in patients with premature adrenarche. J Clin Endocrinol Metab. (1999) 84:1570–4. doi: 10.1210/jc.84.5.1570
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Ambroziak U, Kepczynska-eventi U, Kurylovic, MAŁUNOWICZ EM, Wójcicka, Miscevic P, et al. La diagnosi di iperplasia congenita nonclassic della corteccia surrenale due a 21-hydroxylase deficiency, basato su siero basale o post-ACTH stimulation 17-hydroxyprogesterone, può portare a diagnosi false-positive. Clin Endocrinol (Oxf). (2016) 84:23–9. DOI: 10.1111 / prezzi.12935
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Ambroziak U, Kepczynska-eventi U, Kurylovic, Wysłouch-Cieszyńska a, MAŁUNOWICZ EM, Bartoszewicz, et al. LC-MS/MS improves screening towards 21-hydroxylase deficiency. Gynecol Endocrinol. (2015) 31:296–300. doi: 10.3109/09513590.2014.994599
PubMed Abstract | CrossRef Full Text | Google Scholar
23. BBC, CC, Goldman mm, Zhang K, Clark-N, Reitz re Welt CK. Misurazione simultanea di tredici ormoni steroidei nelle donne con sindrome dell’ovaio policistico e donne di controllo utilizzando cromatografia liquida-spettrometria di massa tandem. PLoS Uno. (2014) 9:e93805. doi: 10.1371 / giornale.pone.0093805
PubMed Abstract | CrossRef Full Text/Google Scholar
24. Ray JA, Kushnir MM, Yost RA, Rockwood AL, Wayne Meikle A. Miglioramento delle prestazioni nella misurazione di 5 steroidi endogeni da LC-MS / MS combinato con spettrometria differenziale di mobilità ionica. Clin Chim Acta. (2015) 438:330–6. doi: 10.1016 / j. cca.2014.07.036
PubMed Abstract | CrossRef Full Text/Google Scholar
25. Stoupa A, González-Briceño L, Pinto G, Samara-Boustani D, Thalassinos C, Flechtner I, et al. Risposta inadeguata del cortisolo al test del tetracosattide (Synacthen®) nell’iperplasia surrenalica congenita non classica: un’eccezione alla regola? Horm Res Paediatr. (2015) 83:262–7. doi: 10.1159/000369901
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. (1992) 90:584–95. doi: 10.1172/JCI115897
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Temeck JW, Pang SY, Nelson C, Nuovo MI. Difetti genetici della steroidogenesi nel pubarca prematuro. J Clin Endocrinol Metab. (1987) 64:609–17. doi: 10.1210 / jcem-64-3-609
PubMed Abstract | CrossRef Full Text/Google Scholar
28. Voutilainen R, Jääskeläinen J. Adrenarche prematuro: eziologia, risultati clinici e conseguenze. J Steroide Biochem Mol Biol. (2015) 145:226–36. doi: 10.1016 / j. jsbmb.2014.06.004
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Oberfield SE, Sopher AB, Gerken AT. Approach to the girl with early onset of pubic hair. J Clin Endocrinol Metab. (2011) 96:1610–22. doi: 10.1210/jc.2011-0225
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Janus D, Wojcik M, Malunowicz E, Starzyk JB. Un caso di aderenze labiali ricorrenti in un bambino di 15 mesi con iperplasia surrenalica congenita asintomatica non classica dovuta a deficit di 21-idrossilasi. J Pediatr Endocrinol Metab. (2012) 25:1017–21. doi: 10.1515/jpem-2012-0190
PubMed Abstract | CrossRef Full Text/Google Scholar
31. Gopal-Kothandapani JS, Petkar A, O’Shea E, Banerjee I. Capelli perianali come una presentazione insolita di iperplasia surrenalica congenita non classica. BMJ Caso Rep. (2013) 2013:bcr2013010123. doi: 10.1136/bcr-2013-010123
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Falhammar H, Nordenström A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine. (2015) 50:32–50. doi: 10.1007/s12020-015-0656-0
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Chrousos GP, Loriaux DL, Mann DL, Cutler GB. Late-onset 21-hydroxylase deficiency mimicking idiopathic hirsutism or polycystic ovarian disease. Ann Intern Med. (1982) 96:143–8. doi: 10.7326/0003-4819-96-2-143
CrossRef Full Text | Google Scholar
34. Witchel SF. Non-classic congenital adrenal hyperplasia. Steroids. (2013) 78:747–50. doi: 10.1016/j.steroids.2013.04.010
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Carmina E, Rosato F, Jannì A, Rizzo M, Longo RA. Extensive clinical experience: relative prevalence of different androgen excess disorders in 950 women referred because of clinical hyperandrogenism. J Clin Endocrinol Metab. (2006) 91:2–6. doi: 10.1210/jc.2005-1457
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Moran C, Azziz R, Weintrob N, Witchel SF, Rohmer V, Dewailly D, et al. Esito riproduttivo di donne con iperplasia surrenalica non classica carente di 21-idrossilasi. J Clin Endocrinol Metab. (2006) 91:3451–6. doi: 10.1210 / cc.2006-0062
PubMed Abstract | CrossRef Full Text/Google Scholar
37. Levin JH, Carmina E, Lobo RA. La secrezione di gonadotropina inappropriata dei pazienti con sindrome dell’ovaio policistico è simile a quella dei pazienti con iperplasia surrenalica congenita ad esordio adulto? Fertil Steril. (1991) 56:635–40. doi: 10.1016/S0015-0282(16)54592-0
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Carmina E, Lobo RA. Ovarian suppression reduces clinical and endocrine expression of late-onset congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Fertil Steril. (1994) 62:738–43. doi: 10.1016/S0015-0282(16)56998-2
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Ludwig E. Classificazione dei tipi di alopecia androgenetica (calvizie comune) che si verificano nel sesso femminile. Br J Dermatol. (1977) 97:247–54. doi: 10.1111 / j. 1365-2133. 1977.tb15179.x
PubMed Abstract | CrossRef Full Text/Google Scholar
40. Trakakis E, Papadavid E, Dalamaga M, Koumaki D, Stavrianeas N, Rigopoulos D, et al. Prevalenza di iperplasia surrenalica congenita non classica dovuta a carenza di 21-idrossilasi nelle donne greche con acne: uno studio trasversale basato sull’ospedale. J Eur Acad Dermatol Venereol. (2013) 27:1448–51. doi: 10.1111 / j. 1468-3083. 2012.04613.x
PubMed Abstract | CrossRef Full Text/Google Scholar
41. I nostri servizi sono a vostra disposizione per ogni esigenza. Stato di salute degli adulti con iperplasia surrenalica congenita: uno studio di coorte su 203 pazienti. J Clin Endocrinol Metab. (2010) 95:5110–21. doi: 10.1210 / cc.2010-0917
PubMed Abstract | CrossRef Full Text/Google Scholar
42. Saygili F, Oge A, Yilmaz C. Iperinsulinemia e insensibilità all’insulina nelle donne con iperplasia surrenalica congenita non classica dovuta a deficit di 21-idrossilasi: la relazione tra i livelli di leptina sierica e iperinsulinemia cronica. Horm Res. (2005) 63:270-4. doi: 10.1159/000086363
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Speiser PW, Serrat J, New MI, Gertner JM. Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1992) 75:1421–4. doi: 10.1210/jc.75.6.1421
PubMed Abstract | CrossRef Full Text | Google Scholar
44. I nostri servizi sono a vostra disposizione. Regime di trattamento con glucocorticoidi e risultati di salute negli adulti con iperplasia surrenalica congenita. Clin Endocrinol. (2013) 78:197–203. doi: 10.1111 / cen.12045
PubMed Abstract | CrossRef Full Text/Google Scholar
45. Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Uso di corticosteroidi orali e rischio di fratture. J Bone Miner Res. (2000) 15:993-1000. doi: 10.1359 / jbmr.2000.15.6.993
PubMed Abstract | CrossRef Full Text | Google Scholar
46. Nebesio TD, Renbarger JL, Nabhan ZM, Ross SE, Slaven JE, Li L, et al. Differential effects of hydrocortisone, prednisone, and dexamethasone on hormonal and pharmacokinetic profiles: a pilot study in children with congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2016) 2016:17. doi: 10.1186 / s13633-016-0035-5
PubMed Abstract | CrossRef Full Text/Google Scholar
47. Weintrob N, Dickerman Z, Sprecher E, Galatzer A, Pertzelan A. Carenza di 21-idrossilasi non classica nell’infanzia e nell’infanzia: l’effetto del tempo di inizio della terapia sulla pubertà e sull’altezza finale. Eur J Endocrinolo. (1997) 136:188–95. doi: 10.1530/eje.0.1360188
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Bonfig W, Bechtold S, Schmidt H, Knorr D, Schwarz HP. Reduced final height outcome in congenital adrenal hyperplasia under prednisone treatment: deceleration of growth velocity during puberty. J Clin Endocrinol Metab. (2007) 92:1635–9. doi: 10.1210/jc.2006-2109
PubMed Abstract | CrossRef Full Text | Google Scholar
49. Dörr HG. Crescita in pazienti con iperplasia surrenalica congenita classica dovuta a deficit di 21-idrossilasi. Horm Res. (2007) 68 (Suppl 5): 93-9. doi: 10.1159 / 000110587
PubMed Abstract | CrossRef Full Text/Google Scholar
50. Hoepffner W, Kaufhold A, Willgerodt H, Keller E. I pazienti con iperplasia surrenalica congenita classica dovuta a carenza di 21-idrossilasi possono raggiungere la loro altezza target: l’esperienza di Lipsia. Horm Res. (2008) 70:42-50. doi: 10.1159/000129677
PubMed Abstract | CrossRef Full Text | Google Scholar
51. van der Kaay D, van den Akker E. Ultralow-dose dexamethasone to preserve endogenous cortisol stress response in nonclassical congenital adrenal hyperplasia: a new promising treatment. Int J Endocrinol Metab. (2014) 12:e14657. doi: 10.5812/ijem.14657
CrossRef Full Text | Google Scholar
52. Lin-Su K, Vogiatzi MG, Marshall I, Harbison MD, Macapagal MC, Betensky B, et al. Treatment with growth hormone and luteinizing hormone releasing hormone analog improves final adult height in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. (2005) 90:3318–25. doi: 10.1210/jc.2004-2128
PubMed Abstract | CrossRef Full Text | Google Scholar
53. McCann-Crosby B, Chen M-J, Lyons SK, Lin Y, Axelrad M, Dietrich JE, et al. Nonclassical congenital adrenal hyperplasia: obiettivi di trattamento e transizione. Pediatr Endocrinol Rev. (2014) 12:224-38.
PubMed Abstract/Google Scholar
54. I nostri servizi sono sempre disponibili. Iperplasia surrenalica congenita negli adulti: una rassegna di problemi medici, chirurgici e psicologici. Clin Endocrinol. (2006) 64:2–11. doi: 10.1111 / j. 1365-2265. 2005.02410.x
PubMed Abstract | CrossRef Full Text | Google Scholar
55. Mah PM, Jenkins RC, Rostami-Hodjegan A, Newell-Price J, Doane A, Ibbotson V, et al. Weight-related dosing, timing and monitoring hydrocortisone replacement therapy in patients with adrenal insufficiency. Clin Endocrinol. (2004) 61:367–75. doi: 10.1111/j.1365-2265.2004.02106.x
PubMed Abstract | CrossRef Full Text | Google Scholar
56. Diaz A, Laufer MR culatta LLAA di PC su Un, Cura AC di O e GC su AH. Mestruazioni nelle ragazze e negli adolescenti: usando il ciclo mestruale come segno vitale. Pediatria. (2006) 118:2245–50. doi: 10.1542 / peds.2006-2481
CrossRef Full Text/Google Scholar
57. Merke DP, Poppas DP. Gestione di adolescenti con iperplasia surrenalica congenita. lancet Diabete Endocrinol. (2013) 1:341–52. doi: 10.1016/S2213-8587(13)70138-4
PubMed Abstract | CrossRef Full Text | Google Scholar
58. Verma S, Green-Golan L, VanRyzin C, Drinkard B, Mehta SP, Weise M, et al. Adrenomedullary function in patients with nonclassic congenital adrenal hyperplasia. Horm Metab Res. (2010) 42:607–12. doi: 10.1055/s-0030-1253385
PubMed Abstract | CrossRef Full Text | Google Scholar
59. New MI. Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2006) 91:4205–14. doi: 10.1210/jc.2006-1645
PubMed Abstract | CrossRef Full Text | Google Scholar
60. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2018) 103:4043–88. doi: 10.1210/jc.2018-01865
PubMed Abstract | CrossRef Full Text | Google Scholar
61. Kruse B, Riepe FG, Krone N, Bosinski HAG, Kloehn S, Partsch CJ, et al. Congenital adrenal hyperplasia – how to improve the transition from adolescence to adult life. Exp Clin Endocrinol Diabetes. (2004) 112:343–55. doi: 10.1055/s-2004-821013
PubMed Abstract | CrossRef Full Text | Google Scholar
62. Gastaud F, Bouvattier C, Duranteau L, Brauner R,Thibaud E, Kutten F, et al. Compromissione dei risultati sessuali e riproduttivi nelle donne con forme classiche di iperplasia surrenalica congenita. J Clin Endocrinol Metab. (2007) 92:1391–6. doi: 10.1210 / cc.2006-1757
PubMed Abstract | CrossRef Full Text/Google Scholar
63. Mercè Fernández-Balsells M, Muthusamy K, Smushkin G, Lampropulos JF, Elamin MB, Abu Elnour NO, et al. Uso prenatale del desametasone per la prevenzione di virilization nelle gravidanze a rischio per iperplasia adrenale congenita classica a causa della carenza di 21-hydroxylase (CYP21A2): una revisione sistematica e meta-analisi. Clin Endocrinol. (2010) 73:436–44. doi: 10.1111 / j. 1365-2265. 2010.03826.x
CrossRef Full Text/Google Scholar
64. Bidet M, Bellanné-Chantelot C, Galand-Portier MB, Golmard JL, Tardy V, Morel Y, et al. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2010) 95:1182–90. doi: 10.1210/jc.2009-1383
PubMed Abstract | CrossRef Full Text | Google Scholar
65. Birnbaum MD, Rose LI. Late onset adrenocortical hydroxylase deficiencies associated with menstrual dysfunction. Obstet Gynecol. (1984) 63:445–51.
PubMed Abstract | Google Scholar
66. Cuhaci N, Aydin C, Yesilyurt A, Pinarli FA, Ersoy R, Cakir B. Nonclassical congenital adrenal hyperplasia and pregnancy. Case Rep Endocrinol. (2015) 2015:296924. doi: 10.1155/2015/296924
PubMed Abstract | CrossRef Full Text | Google Scholar
67. Deneux C, Tardy V, Anne D, Mornet E, Billaud L, Charron D, et al. Correlazione fenotipo-genotipo in 56 donne con iperplasia surrenalica congenita non classica dovuta a deficit di 21-idrossilasi. J Clin Endocrinol Metab. (2001) 86:207–13. doi: 10.1210 / jcem.86.1.7131
PubMed Abstract | CrossRef Full Text/Google Scholar
68. Simonetti L, Bruque CD, Fernández CS, Benavides-Mori B, Delea M, Kolomenski JE, et al. Aggiornamento della mutazione CYP21A2: analisi completa di database e varianti genetiche pubblicate. Ronzio Mutat. (2018) 39:5–22. doi: 10.1002 / humu.23351
PubMed Abstract | CrossRef Full Text | Google Scholar
69. OMIM Entry – * 613815 – CYTOCHROME P450 FAMILY 21 SUBFAMILY A POLYPEPTIDE 2; CYP21A2.
Google Scholar
70. Miller WL, Witchel SF. Prenatal treatment of congenital adrenal hyperplasia: risks outweigh benefits. Am J Obstet Gynecol. (2013) 208:354–9. doi: 10.1016/j.ajog.2012.10.885
PubMed Abstract | CrossRef Full Text | Google Scholar
71. Lekarev O, New MI. Adrenal disease in pregnancy. Best Pract Res Clin Endocrinol Metab. (2011) 25:959–73. doi: 10.1016/j.beem.2011.08.004
PubMed Abstract | CrossRef Full Text | Google Scholar
72. New MI, Abraham M, Yuen T, Lekarev O. An update on prenatal diagnosis and treatment of congenital adrenal hyperplasia. Semin Reprod Med. (2012) 30:396–9. doi: 10.1055/s-0032-1324723
PubMed Abstract | CrossRef Full Text | Google Scholar
73. Kazmi D, Bailey J, Yau M, Abu-Amer W, Kumar A, Low M, et al. New developments in prenatal diagnosis of congenital adrenal hyperplasia. J Steroid Biochem Mol Biol. (2017) 165:121–3. doi: 10.1016/j.jsbmb.2016.06.016
PubMed Abstract | CrossRef Full Text | Google Scholar
74. Clayton PE, Miller WL, Oberfield SE, Ritzén EM, Sippell WG, Speiser PW, et al. Dichiarazione di consenso sulla carenza di 21-idrossilasi dalla Società europea di endocrinologia pediatrica e dalla lawson wilkins pediatric endocrine society. Horm Res. (2002) 58:188-95. doi: 10.1159/000065490
PubMed Abstract | CrossRef Full Text | Google Scholar
75. I nostri servizi sono a vostra disposizione per ogni esigenza. Profilo fenotipico di genitori con iperplasia surrenalica congenita criptica non classica: risultati in 145 famiglie non correlate. Eur J Endocrinolo. (2011) 164:977–84. doi: 10.1530/EJE-11-0019
PubMed Abstract | CrossRef Full Text | Google Scholar
76. Takasu N, Nakachi K, Higa H. Lo sviluppo dell’ipertiroidismo di Graves ha causato una crisi surrenalica in un paziente con deficit di 21-idrossilasi non classico precedentemente non riconosciuto. Stagista Med. (2010) 49:1395–400. doi: 10.2169 / internalmedicina.49.3573
PubMed Abstract | CrossRef Full Text | Google Scholar
77. Meyer-Bahlburg HFL, Dolezal C, Baker SW, Carlson AD, Obeid JS, New MI. Prenatal androgenization affects gender-related behavior but not gender identity in 5-12-year-old girls with congenital adrenal hyperplasia. Arch Sex Behav. (2004) 33:97–104. doi: 10.1023/B:ASEB.0000014324.25718.51
CrossRef Full Text | Google Scholar
78. Meyer-Bahlburg HFL, Dolezal C, Baker SW, Nuova MI. Orientamento sessuale nelle donne con iperplasia surrenalica congenita classica o non classica in funzione del grado di eccesso di androgeni prenatale. Arco Sesso Comportarsi. (2008) 37:85–99. doi: 10.1007 / s10508-007-9265-1
PubMed Abstract | CrossRef Full Text / Google Scholar