- määritelmät ja prevalenssi
- diagnoosi
- Nccah-ilmenemismuodot naisilla
- ilmentymät lapsuudessa
- ilmenemismuodot nuoruusiässä ja aikuisiässä
- Nccah: n hoito ensimmäisestä ilmentymästä aikuisikään
- hoito lapsuudessa
- hoito nuoruusiässä
- lapset
- naiset, joilla ilmenee ensimmäisen kerran nuoruusiän aikana tai Hyperandrogeenisia oireita hoidon lopettamisen jälkeen
- Nccah ja lisääntyminen
- Subfertiliteetti
- keskenmenot
- hedelmällisyyden suunnittelu
- CAH: n Prenataalinen hoito
- hoito raskauden aikana
- muut erityisongelmat
- stressinhallinta nccah: ssa
- Psykobiologiset näkökohdat nccah: ssa
- johtopäätökset
- Tekijäosuudet
- Eturistiriitalausunto
määritelmät ja prevalenssi
synnynnäinen lisämunuaisen hyperplasia (cah) käsittää autosomaalisen resessiivisen häiriöperheen, jolle on ominaista lievä tai akuutisti heikentynyt kortisolin synteesi, joka johtuu kortisolin tuotantoon tarvittavasta viiden lisämunuaisen steroidogeenisen entsyymin puutoksesta (1, 2). Perinteisesti, CAH on jaettu (a) klassinen (CCAH), esittää suolan tuhlausta tai yksinkertainen virilizing muodossa, joka on ilmeinen syntymän ja/tai vastasyntyneen aikana, ja (b) ei-klassinen (NCCAH), joka edustaa vähemmän vakava muoto häiriö, joka ei ole sukupuolielinten epäselvyyttä, ei ole välittömästi hengenvaarallinen, ja esittelee myöhemmin elämässä, tai pysyy oireeton, tai on misdiagnosed kuin eri sairaus (3). CAH: n eri muotojen rajoja ei kuitenkaan ole tarkasti määritelty, mikä on omiaan lisäämään tähän häiriöön liittyviä haasteita. Cah: ta kannattaakin pitää fenotyyppien jatkumona vaikeasta lievään tai muuten oireettomaan (3).
yleisin CAH: n muoto johtuu 21-hydroksylaasin puutteesta (21ohd), mikä johtaa 17 hydroksiprogesteronin (17 OHP) heikentyneeseen tai olemattomaan muuntumiseen 11 deoksikortisoliksi ja progesteronin muuttumiseen deoksikortosteroniksi. Steroidikonversion esto johtaa androgeenien esiasteiden tuotannon lisääntymiseen CRH-ACTH-stimulaatiossa, mikä johtaa biokemialliseen hyperandrogenismiin, jota leimaavat kohonneet 17-OHP-tasot. Taudin klassinen muoto esiintyy 1: llä 16 000: sta elävänä syntyneestä maailmassa. On kuitenkin mainittava, että taudin havaittu esiintyvyys liittyy käytettyyn seulontamenetelmään. Esimerkiksi, se on raportoitu, että esiintyvyys klassisen muodon CAH Ruotsissa on 1:11,500 kun tapaustutkimus lähestymistapa käytetään, mikä luku kuitenkin laskee 1: 9,800 kun hormonaalista seulonta sovelletaan. Samoin esiintyvyys Ranskassa ja Sveitsissä vaihtelee välillä 1:15 472-1:23 000 ja 1:10 749 ja 1:11 661, kun käytetään erilaisia seulontamenetelmiä (4).
Nccah on paljon yleisempi, esiintyy noin yhdellä tuhannesta valkoihoisesta ja yleisemmin tietyissä etnisissä ryhmissä, kuten Ashkenazijuutalaisissa (1:27), latinalaisamerikkalaisissa (1: 53), Jugoslaaveissa (1: 62) ja italialaisissa.(1:300) (5, 6). Eri arviointimenetelmiin (tapaustutkimus, hormoniseulonta ja molekyylitestaus) perustuvia tietoja nccah: n esiintyvyydestä ei kuitenkaan ole. Nccah:ta pidetään yleisimpänä autosomaalisesti resessiivisenä endokriinisenä häiriönä, jonka kantotaajuus on 1:25-1: 10. Nccah: ssa 21-OHD: n vuoksi jäljellä olevan entsymaattisen aktiivisuuden arvioidaan olevan noin 10-70% (7-9).
diagnoosi
verrattuna taudin klassisen muodon diagnoosiin, joka tehdään syntymän tai vastasyntyneen aikana sukupuolielinten epäselvyyden ja / tai suolaa tuhlaavien oireiden vuoksi tai joissakin maissa käytettyjen seulontaohjelmien avulla useimpia nccah-tapauksia ei ole helppo havaita (4, 10). Lisäksi monet yksilöt pysyvät oireettomina lapsuudessa ja nuoruudessa, niillä on normaali lisääntymistoiminta ja ne tulevat tietoisiksi NCCAH: sta vasta toisen perheenjäsenen diagnoosin ja sen seurauksena tehtyjen testien (11) ansiosta. Kuitenkin useimmat naiset, joilla on NCCAH, hakevat lääketieteellistä apua, kun he kokevat androgeeniylimäärän oireita ja kun kliininen epäily vaatii testausta, kohonnut basal 17 OHP-taso todennäköisesti viittaa nccah: n diagnoosiin.
Endocrine Societyn ehdottamissa kliinisissä ohjeissa suositellaan nccah: n seulontavälineeksi 17 OHP: n perusarvoa, jota ei stimuloida. Aamun 17 OHP: n tasot >6 nmol/L follikulaarivaiheessa menstruoivilla naarailla pitäisi vaatia lisäarviointia, koska on osoitettu, että yli 6 nmol/L tasot kuvaavat 90%: a nccah: n yksilöistä (12, 13). Satunnaisista 17 OHP: n mittauksista ei ole ollut hyötyä, koska nccah-potilailla ne tuottavat usein normaaleja pitoisuuksia, ja lisäksi ne ovat erittäin korkeita luteaalivaiheessa puolella terveistä naisista (13). Kuitenkin 280: stä tautia sairastavasta koehenkilöstä saaduissa tiedoissamme kuusi potilasta (2.1%) lähtötason 17 OHP-arvo < 6 nmol/L (14). Lisäksi, Bidet et al., suuressa kohortissa naisia, joilla oli nccah, todettiin molekyylitekniikoilla todennettuna myös 17 OHP: n perusarvot alle 6 nmol/L 8%: lla tutkituista koehenkilöistä (15). Lopuksi Speiserin ym. keräämien tietojen perusteella., 9%: lla nccah-potilaista 17 OHP-arvot olivat alle 2 ng/ml (mikä vastaa 6nmol/L). Muiden tutkimusten mukaan nccah: n (13, 16, 17) diagnosointiin riittää 17 OHP: n lähtöarvo välillä 5, 1-9 nmol/L. Äskettäin taso basal 17 OHP 4.6 nmol/L ehdotettiin kynnysarvoksi ACTH-testille nccah: n ennustamiseksi koehenkilöillä, joilla oli ennenaikainen adrenarche lapsuudessa (18).
näiden löydösten ja ehdotusten yhteismäärä osoittaa, että Synakteenin stimulaatiotestin läpikäyvien potilaiden valinta on arvioitava tapauskohtaisesti. Diagnoosiin tarvitaan 17 OHP: n stimulaation jälkeinen taso yli 3 nmol/L (19). Heterotsygooteilla, joilla on yksi cyp21a2-mutaatio, on ACTH-stimulaation jälkeen hieman koholla 17 OHP-tasoa, joskin päällekkäisyyksiä esiintyy potilailla, joilla ei ole vaikutusta (9). Kuitenkin, kuten Dacou-Voutetakis et al. ovat huomauttaneet, jos perus-ja poststimulaation 17 OHP-arvojen summa ylittää 1,5 nmol/L, heterotsygoottisuuden mahdollisuus on poikkeuksellisen korkea (20).
toinen tärkeä näkökohta liittyy 17 OHP: n arvioinnissa käytettyihin tekniikoihin. 17 OHP-tasoa mitataan erilaisilla immunomääritysmenetelmillä, mutta kuten äskettäin on osoitettu, tarkimmat ja luotettavimmat tulokset saavutettiin nestekromatografian ja massaspektrometrian yhdistelmällä (LC-MS/MS). Itse asiassa monia vääriä positiivisia 17 OHP-mittauksia havaittiin, kun LC-MS/Ms-mittauksia verrattiin standardimenetelmiin (21) kuitenkin jälkimmäisiä menetelmiä ei käytetä yleisesti, jos diagnoosi on epävarma, ne antavat tarkempia tuloksia. On huomattava, että LC-MS: n/MS: n kvantifioitua 17 OHP: tä koskevia seulonta-ja diagnostisia raja-arvoja ei ole vielä määritelty. Nämä kynnysarvot ovat todennäköisesti alhaisemmat kuin immunomäärityksillä vahvistetut, koska LC-MS/MS-menetelmä on spesifisempi, koska se on vähemmän altis ristireaktiivisuudelle ja häiriöille (22, 23). Se, tarvitaanko virtsan steroidiprofiili lopullista diagnoosia varten, on selvitettävä (22). Rajatapauksissa on suositeltavaa saada täydellinen adrenokortinen profiili ACTH-stimulaatiotestin jälkeen 21-hydroksylaasin puutoksen erottamiseksi muista entsyymivirheistä ja vakaan diagnoosin tekemiseksi.
erityisesti epäselvissä tapauksissa on tehtävä täydellinen steroidiprofiili 21-hydroksylaasin puutoksen varmistamiseksi ja muiden entsymaattisten havaintojen poissulkemiseksi. 17 OHP: n, kortisolin, 11-deoksikortisonin,11-deoksikortisolin,17-OH-pregnenolonin, dehydroepiandrosteronin ja androstenedionin sisällyttäminen seerumin steroidiprofiiliin olisi hyödyllistä sulkea pois muut CAH: n syyt, kuten 11β-hydroksylaasin puutos ja harvemmin 3β-hydroksisteroididehydrogenaasin puutos tai P450 oksidoreduktaasin puutos (24). Vaikka geneettistä testausta ei pidetä nccah: n ensisijaisena diagnostisena työkaluna tällä hetkellä, se on pakollinen diagnoosin vahvistamisessa ja geneettisessä neuvonnassa (3).
kortisolitasojen osalta stimulaation jälkeiseksi arvoksi odotetaan yleensä vähintään 496 nmol / L. Huomattava, mukaan Stoupa et al., 60%: lla 47 lapsesta, joilla oli NCCAH 21 OHD: n seurauksena, oli alhaiset kortisoliarvot stimulaation jälkeen, mikä viittaa siihen, että lisämunuaisen vajaatoiminnan kehittymistä on seurattava tehostetusti suurten stressitapahtumien aikana (25).
Nccah-ilmenemismuodot naisilla
nccah: n kliiniselle ilmentymälle on ominaista korkea polymorfisuus, joka liittyy alkamisiän lisäksi myös eri merkkeihin ja oireisiin. Nccah: n ensimmäinen kliininen esiintyminen on raportoitu 11 prosentissa tapauksista ennen 10 vuoden ikää ja 80 prosentissa tapauksista 10-40 vuoden iässä (12). CAH: n ja nccah: n genotyyppi-fenotyyppikorrelaatiota ei ole vielä selvitetty. Speiser ym. viittaa siihen, että suurin osa mutta ei kaikki 21-hydroksylaasin puutoksen fenotyypin vaihtelusta johtuu CYP21A2: n (26) alleelisesta vaihtelusta.
ilmentymät lapsuudessa
vastasyntyneellä tyypillisesti nccah-naaraat pysyvät oireettomina ja niillä on normaalit ulkoiset sukupuolielimet. Varhaisin nccah: n raportoima tapaus on 6 kuukauden ikäinen tyttö, jolle kehittyi häpykarvoja (11). Yleensä kliiniset löydökset ja oireet nccah-tapauksissa alkavat 5 vuoden iästä tai jopa myöhemmin ja liittyvät kohonneeseen androgeenitasoon, vaikka lievää kortisolin puutosta voi esiintyä myös joissakin tapauksissa (8). 92%: ssa nccah-tapauksista ensimmäinen oire on ennenaikainen pubarche (PP), jota esiintyy alle 8-vuotiailla tytöillä ja alle 9-vuotiailla pojilla (12) ja jonka esiintyvyys on arviolta 10-11, 3% (15). Huom. lapsilla, joilla on PP, eri tutkimusten mukaan nccah: n esiintyvyys vaihtelee 5-30%: sta (20, 27). Kuitenkin perustuen analyysiimme 280 potilaasta, joilla oli nccah: n molekyylivahvistus, havaitsimme, että PP: n esiintyvyys 94: llä alle 8-vuotiaalla naisella oli peräti 88%. Toisaalta analyysissä, jossa oli mukana 45 nccah-potilasta, todettiin PP vain 29 prosentissa koehenkilöistä (14). Lisäksi on pidettävä mielessä, että noin puolet PP-potilaista saattaa olla cyp21a2-mutaation heterotsygootteja.
toinen asia, joka kaipaa selvennystä, on PP: n ja varhaiskypsän murrosiän välinen suhde. On oletettu, että joissakin tapauksissa lisämunuaisten androgeenien perifeerinen aromatisaatio estrogeeneihin voi aktivoida hypotalamus-aivolisäke-sukurauhasakselin, mikä johtaa sekundaariseen varhaiskypsään puberteettiin (28). Tällöin tarvitaan säännöllistä seurantaa. Joillekin lapsille kehittyy hirsutismia, aknea ja/tai nopeaa kasvua ja ilmenee pitkä rakenne ja luuston Ikä eteneminen (29). Jälkimmäinen voi johtaa typistetty lopullinen korkeus seurauksena nopea epiphyseal fuusio. Muita harvinaisia kliinisiä löydöksiä lapsuudessa ovat labiaalinen adheesio, perianaalihiukset, klitoromegalia ja äänen käheys (30-32).
ilmenemismuodot nuoruusiässä ja aikuisiässä
nuoruusiässä ja aikuisiässä nccah: lla on naisilla hyperandrogeenisia oireita, jotka muistuttavat munasarjojen monirakkulatautioireyhtymää (PCOS) (33). Oireita ovat hirsutismi, akne, androgeeninen hiustenlähtö, anovulaatio, kuukautishäiriöt ja hedelmättömyys. Monikeskustutkimuksessa yleisimpiä oireita nuorilla ja aikuisilla naisilla olivat hirsutismi (59%), oligomenorrea (54%) ja akne (33%). Nccah-tautia sairastavista 161 naisesta oireina olivat hirsutismi (78%), kuukautishäiriöt (54, 7%) ja alentunut hedelmällisyys (12%) (34). Ryhmässämme 161 naista, joilla oli tauti 63%: lla potilaista, joilla oli munasarjojen monirakkulatyyppinen fenotyyppi (14). Tällä löydöksellä on toisinpäin vaikutus, sillä nccah: n esiintyvyys on korkea naisilla, joilla on hyperandrogenemia. Varmasti, tutkimus Witcel et al. nccah: n ilmaantuvuuden raportointi 1-33% hyperandrogeenisillä naisilla on erittäin kiinnostavaa. Toisessa tutkimuksessa 950 naista, jotka lähetettiin androgeeniylimäärän arviointiin, nccah havaittiin 4,2%: lla heistä. Nccah-potilaat olivat kuitenkin nuorempia ja hirsutiilisempia verrattuna muihin hyperandrogeenisiin potilasryhmiin. Heille oli myös ominaista huomattavasti korkeampi testosteronitaso, vapaa testosteroni ja 17 OHP kuin potilailla, joilla oli muita hyperandrogeenisia oireyhtymiä. Munasarjojen ultraäänitutkimuksessa niistä 77 prosentilla näkyi munasarjojen monirakkulatauti ja 41 prosentilla suurentunut munasarjojen koko. Ne täyttivät NIH: n mukaan PCOS-kriteerit 56 ja Rotterdam 72,8 prosentin osuudella (35).
taudin etenevyyttä korostaa se, että hirsutismin esiintyvyyden on osoitettu lisääntyvän iän myötä ja sen on havaittu olevan harvinaista ennen murrosikää. Hirsutismi on yleisin nccah-potilailla raportoitu kliininen ilmentymä, joka vaihtelee 71-96%: lla (28, 36). Puberteetti tytöt nccah tyypillisesti läsnä hirsutismi (37, 38). Toisessa ääripäässä on hiustenlähtö. Miesten kaljuuntumista on raportoitu NCCAH-potilailla. Alopekian esiintyvyys näytti myös lisääntyvän iän myötä 6 prosentista toisella vuosikymmenellä 19 prosenttiin viidennellä vuosikymmenellä, mikä osoittaa jälleen taudin etenevän (39). Aknea on todettu lähes 33%: lla nccah-koehenkilöistä (36). Huomattavaa on, että nccah: ta aiheuttavan CYP21A2-geenin mutaatioita havaittiin 4, 9%: lla 123 aikuisesta naisesta, joilla oli vaikea akne (40).
kuukautiskierron väärinkäytöksiä mukaan lukien oligomenorrea tai toissijainen kuukautisia voi usein olla esillä merkki NCCAH post menarche yksilöitä. Lisäksi 8-9%: ssa tapauksista esiintyy myös primaarista kuukautisia ja tästä syystä hakeudutaan ensimmäistä kertaa lääkäriin. Lisäksi keskuudessa NCCAH yksilöiden, oligomenorrea on yleisempää kuin ensisijainen kuukautisia. Esimerkiksi sarjassa Moran et al., keskuudessa nccah vuotiaiden naisten 10-19 vuotta, oligomenorrea oli läsnä 56% tapauksista verrattuna vain 9% nuoruusiässä ensisijainen kuukautisia (12). Lopuksi, niin monet nccah oireet muistuttavat PCOS, että ei ole yllättävää, että NCCAH on kutsuttu suuri matkija PCOS. Joka tapauksessa, diagnoosi nccah olisi harkittava arvioinnin aikana jokainen nuori nainen, joka on tarkoitettu hyperandrogeenisten oireiden.
Arlt et al: n tutkimuksessa. nccah-potilailla oli merkitsevästi korkeampi painoindeksi ja pienempi lopullinen korkeus verrattuna verrokkeihin (41). Nämä tiedot yhdessä muutamien insuliiniherkkyyteen viittaavien tutkimusten kanssa viittaavat epäsuotuisaan metaboliseen profiiliin, joka voi olla patofysiologisesti selitettävissä kohonneiden androgeenipitoisuuksien vaikutuksella ja/tai glukokortikoidihoidon (42, 43) seurauksena. Osteoporoosi voidaan havaita myös näillä henkilöillä, todennäköisesti kortikosteroidihoidon seurauksena (34).
Nccah: n hoito ensimmäisestä ilmentymästä aikuisikään
kun nccah: n diagnoosi on varmistettu, useita myöhempiin hoitosuosituksiin liittyviä seikkoja on syytä harkita. Ensimmäinen käsiteltävä kysymys on, onko glukokortikoidihoito indisoitu. Tämän lähestymistavan yleinen perustelu on, että tarjoamalla potilaalle riittävät kortisolipitoisuudet, hänen päivittäiset tarpeensa katetaan ja näin ollen CRH-ACTH-akselin stimulaatio kapenee, mikä johtaa lisämunuaisen androgeenituotannon vähenemiseen. Tätä lähestymistapaa koskeva yleissääntö on, että GCs annetaan korvausannoksilla eikä farmakologisilla annoksilla, kun taas iän, sukupuolen, laboratoriotulosten, potilaskohtaisten suositusten ja hoidon tavoitteiden vaikutus glukokortikoidikorvaushoitoon otetaan vakavasti huomioon. Meidän tulisi kuitenkin myös olla tietoisia siitä, että pitkäaikainen GC-korvaushoito voi johtaa hyperkortisolemiaan, josta seuraa hyvin dokumentoituja haittavaikutuksia aineenvaihdunnan kaikkiin osa-alueisiin, erityisesti kasvuun, rasvan jakautumiseen ja insuliiniresistenssiin sekä psykologiseen profiiliin. Toinen merkittävä haitta tässä lähestymistavassa on riittävän kliinisen indeksin tai riittävän korvausannoksen biokemiallisen merkkiaineen puute, kuten TSH-arvojen suhteen kilpirauhasen vajaatoiminnassa.
lopuksi se, että ei ole yksimielisyyttä siitä, mitä GC: tä tulisi käyttää, ja pitkäaikaisen tiedon puuttuminen erilaisista GC: n antotavoista vaikeuttavat hoitavan lääkärin päätöksiä kunkin potilaan optimaalisesta valinnasta ja valintaa. Hydrokortisonia käytetään tyypillisesti lapsilla, koska se muistuttaa eniten luonnollista hormonia (kortisolia), mutta sitä ei pidetä sopivana lähestymistapana nuorilla ja nuorilla naisilla, koska tarvitaan useita vuorokausiannoksia. Siksi useimmat aikuisten endokrinologit suosivat joko deksametasonia tai prednisolonia sopivina annoksina. Toisaalta on korostettava, että eri GCs: ien vastaavuus perustuu niiden anti-inflammatoriseen vaikutukseen eikä ihmisen aineenvaihdunnan eri puoliin. Koska kortisoli vaikuttaa lähes 20 prosenttiin ihmisen perimästä, eri kudoksissa odotetaan erilaisia GCs-vasteita. Tätä käsitystä tukee se, että deksametasonin antaminen CAH: ta sairastaville potilaille on osoittanut insuliiniresistenssin indeksien heikkenemistä muihin GCs: ään verrattuna (44). Lisäksi 2, 5-7, 5 mg: n prednisoloniannoksella, jota pidetään normaalina annoksena, on pitkäaikainen negatiivinen vaikutus luun aineenvaihduntaan (45). Siksi hydrokortisonia on pidettävä parhaana hoitomuotona, jos potilaalle annetaan GC-täydennyshoitoa. Uuden syntetisoidun hydrokortisonikaavan tulo yhdellä pillerillä päivässä ja sen alustavat positiiviset tulokset Cah-potilailla osoittavat paljon lupausta tulevaisuuteen (46).
hoito lapsuudessa
nccah-potilaita, joilla on lapsuudessa diagnosoitu PP: n oireita, voidaan hoitaa hydrokortisonilla lisämunuaishormonien vaimentamiseksi ja luuston iän nopean etenemisen estämiseksi, mikä voi vaikuttaa lopulliseen pituuteen. Niillä potilailla, joilla hydrokortisonihoito aloitettiin 1 vuotta ennen murrosiän alkamista ja joiden luusto-ikä oli alle 9 vuotta, lopullinen pituus pysyi geneettisen potentiaalin rajoissa (47). Käytettävissä olevat tutkimukset osoittavat, että aikuisten pituus lähestyi odotettua tavoitekorkeutta potilailla, joita seurattiin tarkasti ja jotka noudattivat tiukasti lääkityssuunnitelmia (48-50). Äskettäinen pieni tutkimus, jossa oli mukana viisi tapausta (6, 1–9, 2-vuotiaat potilaat), osoitti, että erittäin pieni annos deksametasonia on lupaava vaihtoehto endogeenisen stressin ja sen vaikutusten vähentämiseksi. Onko tämä realistinen lähestymistapa ja kuinka paljon annettua annosta tulisi edelleen selvittää (51). Tutkimus Nebesio et al. verrattiin hydrokortisonin, prednisolonin ja deksametasonin hormonaalisia vaikutuksia ja farmakokinetiikkaa 9 prepubertaalipotilaalla, joilla oli CCAH. Ne osoittivat, että deksametasoni oli voimakkaampi kuin muut muodot saavuttaessaan merkittävästi alhaisemmat lisämunuaisen hormonitasot, mikä viittaa siihen, että deksametasoni on tehokkaampi lisämunuaisen androgeenituotannon tukahduttamisessa (46). Tämän havainnon todentamiseksi tai hylkäämiseksi tarvitaan tietenkin lisätutkimuksia. Lisäksi joissakin tapauksissa kohonneet androgeenipitoisuudet voivat johtaa GnRH-akselin sekundaariseen stimulaatioon, mikä johtaa ennenaikaiseen puberteettiin. Tällöin voidaan määrätä rinnakkaiskurssi GnRH-analogin kanssa, jos luun ikä on huomattavasti korkeampi kuin kronologinen ikä ja/tai ennustettu lopullinen korkeus on suhteeton tavoitekorkeuteen nähden.
toinen tärkeä näkökohta koskee samanaikaista GH: n puutosta silloin, kun GnRH-analogeilla ja GH: n reseptillä saavutettu tavoitekorkeus saavutettiin (52). Nykysuositusten mukaan edellä kuvattua hoitoa suositellaan varovaisesti vain niissä tapauksissa, joissa ennustettu pituuden SD on -2, 25 < tavoitekorkeus tai jopa alempi (53). Vaihtoehtoinen käyttöaihe hydrokortisonihoidon aloittamiselle on riittämätön KORTISOLIVASTE ACTH-stimulaation jälkeen (36).
ensisijainen GC-hoito lapsilla on yleensä hydrokortisoni 10-15 mg / m2 jaettuna kolmeen annokseen. Usein myös pienemmät annokset ovat osoittautuneet tehokkaiksi, alkaen 6 mg/m2 / vrk (34). Yliannostusta tulisi välttää, koska se voi johtaa heikkoon kasvuun sekä Cushingoid-ominaisuuksiin. Säännöllinen kasvutapa, kronologiseen ikään sopiva luun ikä ja keskeisen lihavuuden puuttuminen voivat myös toimia kliinisinä indekseinä asianmukaisen hoidon kannalta. Biokemiallisen / hormonaalisen profiilin osalta androstenedioni-ja testosteronitasoja luuiän Keski-ja yläosissa pidetään parempina merkkiaineina riittävästä GC-korvaushoidosta lapsilla, joilla on NCCAH. Testosteronitasojen suppressiota havaittiin kuitenkin 10%: lla nccah-potilaista, kun taas 28%: lla potilaista testosteronipitoisuudet olivat kohonneet, mikä johtui mahdollisesti hydrokortisonin vaihtelusta (54, 55). DHEAS-arvot pienenevät hyvin pienillä annoksilla, kun taas 17 OHP: n ja progesteronitason tukahduttaminen vaatii erittäin suuria GC-annoksia. On huomattava, että 17 OHP–arvot ovat ratkaisevia diagnoosin kannalta, mutta eivät auta seurannan aikana. Huomattava, verinäytteenotto hormonaalista arviointia varten on suoritettava lopettamatta hoitoa. Sairauden hämmennyksen ja sen monitahoisen luonteen vuoksi ei kuitenkaan ole erityisiä ohjeita hoito-ohjelmien muutosten tai glukokortikoidihoidon lopettamisen ajoitukselle lapsilla.
hoito nuoruusiässä
lapset
kunnes normaali kuukautiskuvio nccah-tytöillä vakiintuu, on erittäin suositeltavaa jatkaa lapsuudessa alkanutta GCs: ää (56). Nuoret potilaat eivät kuitenkaan useinkaan ole riittävän yhdenmukaisia pitkäaikaisen lääkityksen kanssa ja jättävät usein annoksia ottamatta. Lisäksi murrosiässä hydrokortisonin puoliintumisaika laskee 50% lisääntyneen IGF-1-tason seurauksena, mikä vähentää 11ßOHSD-aktiivisuutta, sekä lisääntyneen kortisolin puhdistuman vuoksi, joka johtuu glomerulussuodatusnopeuden (57) monistumisesta. Vuonna nuori nainen, 2 vuotta post menarche ja jos normaali ovulaatio syklit on kirjattu, potilaskeskeinen lähestymistapa kohti hyperandrogeenisia oireita, jotka voivat näkyä on erittäin suositeltavaa.
Jos ilmenee vakavia hyperandrogeenisiä löydöksiä, kuten hirsutismia, aknea ja / tai oligomenorreaa, hoidon jatkamista harkitaan. On tärkeää muistaa nuoren nuoren hauraat tunteet, joita hyperandrogenismin ”hylkivät” merkit vahingoittavat vakavasti. Hyperandrogeenisten merkkien, kuukautishäiriöiden ja huonon elämänlaadun yhdistelmä on hyvin dokumentoitu ja se on erityisen korkea nuoremmilla naisilla. Toinen huomioon otettava seikka on adrenal reserve post ACTH-stimulaation aiheuttama vaste. Koska puberteiinin ja aikuisten naisten kortisolin huippu stimulaation jälkeen on alle 496 nmol / L, stressitapauksissa on annettava steroidihoitoa (58). Toisaalta, jos mitään edellä mainituista oireista ei ilmene nuorella potilaalla, GC-hoito voidaan lopettaa, minkä jälkeen suositellaan säännöllistä seurantaa.
naiset, joilla ilmenee ensimmäisen kerran nuoruusiän aikana tai Hyperandrogeenisia oireita hoidon lopettamisen jälkeen
hyperandrogeenisten oireiden esiintyessä on erittäin suositeltavaa soveltaa potilaslähtöistä lähestymistapaa, jossa keskitytään potilaan pääasiallisiin vaivoihin. Tämä on paljon parempi kuin yleinen yleinen toimintatapa, koska viimeksi mainitussa lähestymistavassa nuori potilas joutuu pettymään, mikä johtaa hoidon keskeyttämiseen ja sen seurauksena epäedullisiin terveysvaikutuksiin aikuisiällä. Tapauksissa, joissa on vaikea hirsutismi, 6-12 kuukauden kurssi suun kautta otettavilla ehkäisypillereillä (OCPs) on kohtuullinen menettely, kun otetaan huomioon OCPs: n suotuisat vaikutukset SHBG-maksan tuotantoon ja androgeenien vapautumisen väheneminen munasarjoista. Kliinistä paranemista odotetaan vähintään 2-3 kuukauden OCPs-hoidon aloittamisen jälkeen, kun taas progestageenin, jolla on vähän androgeenisia ominaisuuksia, samanaikainen käyttö on erittäin suositeltavaa. Jos OCPs epäonnistuu ensimmäisen linjan lähestymistavassa, antiandrogeenit (spironolaktoni, Flutamidi, bikalutamidi, syproteroniasetaatti ja finasteridi) voidaan lisätä hoitoon. Kokemuksemme mukaan bikalutamidin anto on saavuttanut merkittävää parannusta vaikeissa aknetapauksissa, mutta samanlaisia tuloksia ei saatu tapauksissa, joissa on vaikea hirsutismi. Kosmeettisia lähestymistapoja, kuten laser sovellus ja depilatories voidaan myös ehdottaa naisille valittavat liiallinen tai ei-toivottu hiusten kasvua tai päänahan hiustenlähtö (androgeeni hiustenlähtö) (32, 34, 57).
potilaille, joilla on riittämätön kortisolin eritys stimulaation jälkeen, tai jos OKP: tä ja antiandrogeeneja ei voida sietää, GCS-hoito on erittäin suositeltavaa (57). Tiedot New et al. epäsäännölliset kuukautiset ja akne korjaantuvat 3 kuukauden kuluessa glukokortikoidihoidon aloittamisesta, kun taas hirsutismi vaatii lähes 30 kuukautta (59). Toisin kuin lapsuudessa, nuoruusiässä käytetään usein pitempivaikutteisia steroideja ja suositellaan 5 mg prednisolonia tai 0, 25 mg deksametasonia (53). Reaalimaailmassa kliiniset tiedot ovat kuitenkin osoittaneet useita erilaisia nccah-hoidossa sovellettavia hoito-ohjelmia (41).
hoidon ensisijaisena tavoitteena tulee olla potilaan hyperandrogeenisten oireiden lievittäminen. Näin ollen niillä henkilöillä, joilla nccah on diagnosoitu yksinomaan laboratoriotulosten perusteella, kliinisten ominaisuuksien tulisi ohjata hoitosuosituksia, koska hormonaaliset ja molekulaariset löydökset eivät välttämättä ennusta kliinistä vaikeusastetta. Endocrine Societyn ohjeiden mukaan nccah-potilaille tulisi antaa mahdollisuus keskeyttää GC-hoito, kun oireet häviävät (60). Näitä potilaita ei tietenkään pidä hukata seurantaan, vaan hoito tulee aloittaa uudelleen oireiden uusiutuessa. Jos hoito lopetetaan, potilaille on tiedotettava hedelmättömyyden mahdollisuudesta ja heitä on kannustettava ottamaan yhteyttä lääkäriin, jos he haluavat tulla raskaaksi (19). On huomattava, että potilaan asianmukainen siirto lapsipotilaalta aikuiselle endokrinologille on suoritettava optimaalisesti 1 vuoden synkronoidun seurannan jälkeen (53, 61).
Nccah ja lisääntyminen
Subfertiliteetti
Subfertiliteetti on yleisesti raportoitu CAH: n klassisissa muodoissa, mikä johtuu pääasiassa kuukautishäiriöistä, kroonisesta anovulaatiosta ja anatomisista epämuodostumista. Syntyvyydeksi on arvioitu 17 prosenttia, kun verrokkiväestön osuus oli 65 prosenttia (62). Samaan aikaan tiedot hedelmällisyydestä naisilla nccah on viime aikoina arvioitu yksityiskohtaisesti ja arvioitu hedelmättömyys ilmaantuvuus on 11% nccah naisten, eli suhteellisen lievempi kuin Cah, ja monissa tapauksissa on helppo ratkaista. Bidet ym. arvioitiin hedelmällisyyttä 190: llä nccah-tautia sairastavalla naisella, joista 95 halusi tulla raskaaksi. Tässä populaatiossa raportoitiin 187 raskautta 85 naisesta, mikä johti 141 synnytykseen 82 yksilöstä. On korostettava, että 99 raskauksista (52,9%) tapahtui ennen nccah: n diagnoosia, kolme niistä soveltamalla ovulaatioinduktioprotokollia, loput ovat spontaaneja, kun taas 88 raskautta tapahtui nccah: n diagnoosin jälkeen. Valtaosassa niistä raskaus kehittyi hydrokortisonin hoidon jälkeen, kun taas 11 naisella se tapahtui spontaanisti (63, 64). Toisessa raportissa 22: sta nccah: n naisesta, jotka halusivat raskautta, 12 raskautta seurasi prednisoni (65).
keskenmenot
bidet et al-ryhmän kohortissa. GCs: llä hoidetuilla potilailla keskenmenoja oli 6, 5% ja hoitamattomilla potilailla 26, 3% (64). Raskauden lopputulos voi jopa onnistua ilman glukokortikoidihoitoa tapauksissa, joissa nccahia ei ole vielä diagnosoitu, kuten myös cuhaci et al: n tapausraportissa kerrottiin. Tästä huolimatta sama pariskunta koki kaksi keskenmenoa ja ilmoitti olevansa alakuloinen tämän ensimmäisen raskauden jälkeen (66). Bidet ym. ovat arvioineet ennenaikaisten raskauksien mahdollisuutta., se ei eroa merkittävästi muusta väestöstä (15).
hedelmällisyyden suunnittelu
potilaille, joilla on oireinen hyperandrogenismi tai raportoitu lapsettomuus mutta jotka haluavat tulla raskaaksi, suositellaan GC-hoitoa. Tänä aikana käytettävät glukokortikoidit ovat prednisoni tai hydrokortisoni. Tärkein toimi, koska se ohjaa myöhemmät vaiheet, on geneettinen testaus mahdollisten vanhempien. Naaraat, joilla on nccah, jotka haluavat tulla raskaaksi, ovat tietoisia riskistä synnyttää lapsen, jolla on taudin klassinen muoto. Nccah: ta sairastavien naisten raskaustuloksen ja tarkemmin sanottuna CCAH: ta sairastavien lasten esiintyvyyden on arvioitu viimeaikaisissa tutkimuksissa olevan 1, 5-2, 5%, nccah: n ollessa noin 15% (36, 64). Tämä yleisyys riippuu tietysti viiteväestöstä, sillä se on yleisempää väestössä, jossa on paljon seka-avioliittoja (36). Ennustettu todennäköisyys, että vanhemmat synnyttävät lapsen, jolla on Cah, on 1: 120 tuntemattoman isän genotyypin osalta, nolla, jos isällä ei ole asiaankuuluvaa mutaatiota, ja 1: 4, Jos hänellä on heterotsygoottinen mutaatio (19). Tämän seurauksena näiden naisten kumppaneiden geneettinen testaus on välttämätöntä, jotta voidaan arvioida riskiä synnyttää lapsi, jolla on klassinen muoto CAH (64, 67).
siinä tapauksessa, että tulevalla äidillä on kaksi NC-mutaatiota (Taulukko 1), tulevalla isällä ei ole ehdotonta tarvetta geenitestille, sillä jos hän on cyp12a2-mutaatioiden suhteen heterotsygoottinen, jälkeläisillä on riski sairastua taudin ei-klassiseen muotoon tulevaisuudessa. On kuitenkin huomattava, että kantajuus on 2-15% eri populaatioissa ja puolet näistä yksilöistä kantaa vakavaa mutaatiota. Lisäksi lopulliset suositukset tilanteista, joissa geneettistä testausta ei vaadita, ovat vaikeita, koska genotyyppi-fenotyyppi-korrelaatio on epätäydellinen, erityisesti lievempien mutaatioiden osalta . Esimerkiksi P30L-mutaatio, joka useimmiten liittyy nccah: iin, esiintyy myös potilailla, joilla on klassinen CAH . Hiljattain tehty laaja eurooppalainen monikeskustutkimus osoitti, että näin oli 78 prosentilla potilaista . Tässä tutkimuksessa lievä V218L-mutaatio liittyi Nccah: n sijaan klassiseen 30 prosentissa tapauksista. Siksi kaikille nccah-potilaille tulisi tarjota geneettistä neuvontaa ja lisääntymiskumppaneiden molekyyliarviointia.
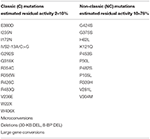
Taulukko 1. Cyp21a2: n mutaatiot, joissa on vähäinen (C) tai kohtalainen (NC) entsyymiaktiivisuus (14, 68, 69).
sen sijaan siinä tapauksessa, että tuleva äiti on heterotsygoottiyhdiste, jolla on yksi vakava (C) ja yksi NC-mutaatio, tulevan isän geneettinen testaus on pakollista. Meidän on pidettävä mielessä, että tiedot, jotka ovat peräisin tutkimuksista, joissa käytetään immunomäärityksiä tai tarkempia LC-MS/MS-menetelmiä, korostavat johdonmukaisesti kyvyttömyyttä luotettavasti sulkea pois heterotsygositeetti käyttämällä basal-tai ACTH-stimuloituja 17 OHP-arvoja, koska ne ovat merkittävästi päällekkäisiä normaalin alueen kanssa. Voidaan ehdottaa Synakteenitestin käyttöä, ja jos se ei ole yhteensopiva heterotsygositeetin kanssa (tyvitumakkeen ja stimuloidun huippuarvon summa 17 OHP < 1, 5 nmol/L), DNA-testiä voidaan välttää. Meidän pitäisi kuitenkin olla hyvin varovaisia tämän testin tulkinnassa, koska sitä ei ole vahvistettu muissa tutkimuksissa. Näin ollen ACTH-stimuloidun 21-deoksikortisolin käyttö joko yksittäisenä tai osana steroidisuhdetta tai steroidiprofiilia voi helpottaa heterotsygoottien biokemiallista tunnistamista tulevaisuudessa, erityisesti LC-MS/MS: n yleistyessä . Jos tuleva isä kantaa NC-mutaatiota, mitään muuta ei tarvita. Päinvastoin, jos hän on klassisen mutaation kantaja, sikiön raskaudenaikaista hoitoa koskeva kysymys nousee esiin. Kaikki mahdolliset genotyyppiyhdistelmät ja ehdotetut menettelyt on esitetty taulukossa 2.

Taulukko 2. Suunniteltujen toimenpiteiden suunnittelu raskauden aikana mahdollisten vanhempien genotyypin perusteella.
CAH: n Prenataalinen hoito
CAH: n prenataalisen hoidon tavoitteena on ehkäistä sikiön genitaaliviriloitumista, mutta myös lievittää stressiä, jota tuntevat vanhemmat, joilla on todennäköisesti monitulkintainen sukuelin (70). Deksametasonia käytetään, koska se läpäisee istukan ja koska se ei deaktivoidu istukan 11β OH-steroididehydrogenaasista ja sitoutuu vain vähän äidin veren kortisolia sitovaan globuliiniin (CBG). Deksametasoni vaimentaa sikiön ACTH-eritystä ja vähentää kohonnutta sikiön androgeenituotantoa. Hoito on keskeytettävä, jos karyotyyppi-tai DNA-analyysi paljastaa miehen tai naisen, jolla ei ole vaikutusta (71). Kuitenkin, vaikka sikiön sukupuolielinten virilization on jo alkanut 6-7 viikkoa hedelmöityksen jälkeen, korioninen villous koepaloja geneettisen diagnoosin ei voida saada ennen 10. -12. viikolla. Tämä aikaväli viittaa siihen, että kaikki raskaudet riski virilizing CAH tulisi hoitaa, vaikka vain 1/8 sikiöt vaikuttaa ja naisten (19, 34, 70).
on edelleen kiistanalaista, onko tämä sikiön altistuminen deksametasonille ennaltaehkäisevissä toimenpiteissä lääketieteellisesti ja eettisesti hyväksyttävää (70). Eläinkokeet ovat osoittaneet, että ensimmäisen raskauskolmanneksen deksametasoni vähentää syntymäpainoa, vaikuttaa munuaisten haiman beetasolujen toimintaan ja aivojen kehitykseen sekä altistaa ahdistukselle, verenpaineelle ja hyperglykemialle aikuisiällä (70). Lisäksi 10-20% raskaana olevista naisista, jotka käyttävät deksametasonihoitoa raskauden aikana, valittavat painonnoususta, ruokahalun lisääntymisestä, mielialan vaihteluista, unettomuudesta, turvotuksesta, verenpaineesta, hyperglykemiasta ja juovista. Kuitenkin Uusi et al. havaittiin keskimääräistä alhaisempi Prader-pistemäärä sikiöillä, joita hoidettiin deksametasonilla ennen syntymää, mutta ei eroa pitkän aikavälin tuloksissa (72). Lisäksi arvostelussa Merce Fernandez-Balsells et al. deksametasonin on osoitettu olevan yhteydessä sikiön virilisaation vähenemiseen ilman merkittäviä äitiin tai sikiöön kohdistuvia haittavaikutuksia (63). Edellä mainitun katsauksen sekä endokrinologisen seuran perättäisten ohjeiden tekijät kuitenkin huomauttavat, että koko kirjallisuuden pienen otoskoon vuoksi aihe on edelleen epävarma ja lisätutkimuksia tarvitaan selvästi.
päätös hoidon aloittamisesta tulee tehdä vain suurissa keskuksissa, joissa on kokenut tiimi ja instituutioiden Arviointilautakuntien hyväksymät protokollat ja jotka perustuvat perheen arvoihin ja mieltymyksiin sekä edellytyksenä heidän kirjalliseen tietoon perustuvaan suostumukseensa (19, 63). Useat alan asiantuntijat ovat sitä mieltä, että olisi kiinnitettävä enemmän huomiota siihen, ettei äiti ja sikiö altistu tarpeettomasti deksametasonille ennen syntymää, sen sijaan että vanhemmat ja potilaat sälyttäisivät moniselitteisten sukupuolielinten tunneperäisen veronsa. Mikä tärkeintä, vanhemmille olisi selvennettävä, että deksametasonin anto ei muuta potilaan asemaa vaan tähtää leikkaustarpeen vähentämiseen eikä elämän tai älyllisen kapasiteetin säilyttämiseen. Heidän on myös tiedettävä, että todennäköisyys, että heidän lapsensa kärsii taudin klassisesta muodosta, on suuri hoidosta huolimatta. Ratkaisevinta tällä hetkellä on, että vankat ja kiistattomat tiedot eivät tue nuoren organismin altistumista erittäin voimakkaalle GC: lle erityisen herkkänä sikiön ohjelmoinnin ja kasvun aikana, mikä saattaa hyvinkin osoittautua hyödyttömäksi XY-karyotyyppisen sikiön tapauksessa.
Ihannetapauksessa diagnoosi olisi tehtävä varhaisessa vaiheessa, ja se olisi tehtävä ei-invasiivisella menetelmällä ja ennen sikiön genitaalivirilisaation alkua. Tähän suuntaan toimivat uudet tutkimukset viittaavat äidin plasmasta saadun soluttoman sikiön DNA: n käyttöön lupaavana menetelmänä, joka mahdollistaa sikiön sukupuolen määrittämisen ja CAH: n diagnosoinnin varhaisessa raskausiässä (<9 viikkoa). Jos tämä menettely toteutetaan laajalti kliinisessä käytännössä, vältetään tarpeetonta prenataalista deksametasonihoitoa (73).
hoito raskauden aikana
raskauden aikana nccah: ta sairastavia naisia tulisi mieluiten hoitaa hydrokortisonilla, ei vain siksi, että raskauteen tyypillisesti liittyy kohonnut stressitaso, vaan myös ennalta ehkäisevänä toimenpiteenä edellä mainittua suurta keskenmenojen esiintyvyyttä vastaan. Tätä glukokortikoidimuotoa suositaan, koska se metaboloituu istukan entsyymin 11β OH-steroididehydrogenaasi II: n avulla, mikä estää glukokortikoidia vaikuttamasta sikiöön (71). Hedelmöityshetkellä hydrokortisoniannos on nostettava 20-25 mg: aan/vrk ja annosmuutokset tehdään 6-8 viikon välein, jotta testosteronitasot pysyisivät normaalin raskauden raskauskolmanneksen (74) ylimmillä tasoilla. Esimerkiksi nccah-potilaan testosteroni-ja D4-arvot hedelmöityksestä jopa 3 vuotta kahden terveen kaksosen synnytyksen jälkeen (kuva 1).
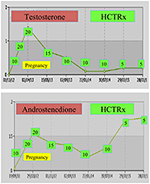
kuva 1. Testosteroni – ja andostenedionitasot ja myöhempi hydrokortisoniannos (HCTRx), hedelmöityksestä 3 vuoden kuluttua nccah-potilaalle annetusta synnytyksestä.
muut erityisongelmat
stressinhallinta nccah: ssa
vakavan hengenvaarallisen stressin, leikkauksen tai vakavan sairauden aikana glukokortikoidihoitoa saavat nccah-potilaat saattavat tarvita suurempia tai useammin glukokortikoidiannoksia, koska heidän lisämunuaisen toiminta on iatrogeenisesti vaimentunut. Siksi on ratkaisevan tärkeää valistaa pienten lasten vanhempia sekä uudelleen valistaa potilaita heidän siirtymisestään aikuisten hoitoon, stressin annostelusta. Jos Nccah-potilaita ei hoideta GCs: llä tai jos hoito lopetetaan, tulee arvioida KORTISOLIVASTE ACTH: lle. Lähes kolmannes nccah-potilaista reagoi stimulaatioon riittämättömästi (15, 75). Niille, joilla ACTH-stimulaatio tehoaa normaalisti, ei suositella stressiannosten hoitoa (19). Mineralokortikoidihoitoa ei tarvita missään näistä tapauksista, koska näillä potilailla aldosteronin eritys on normaalia eikä suolanhukkaa kehity (58). Huomaa, Jos kyseessä on kilpirauhasen liikatoiminta tai levotyroksiinihoidon lisääntyminen, kortisolin puhdistuma lisääntyy ja lisämunuaiskriisi voi esiintyä (58, 76).
Psykobiologiset näkökohdat nccah: ssa
Meyer-Bahlburg et al. ovat useissa tutkimuksissa ilmoittaneet nccah-potilaiden psykologisen profiilin heikentyneen 21OH-puutoksen vuoksi. Tarkemmin, vaikuttaa naisilla oli korkeampi masculinization/defeminization pisteet useita toimenpiteitä sukupuoleen liittyvää käyttäytymistä verrattuna normaaliin verrokki naiset, vaikka huomattavasti vähemmän kuin naisilla klassisen CAH. Heillä oli myös merkittävästi lisääntynyt biseksuaalinen tai homoseksuaalinen suuntautuminen. Retrospektiiviset kliiniset-laadulliset haastattelut näiden naisten kanssa paljastivat epämukavuutta ja sosiaalista stressiä, jotka liittyvät heidän hoidon edeltäviin kokemuksiin androgeeniriippuvaisista oireista, kuten aknesta, hirsutismista ja hedelmöitysvaikeuksista (59, 77, 78). Samoin Arlt et al. osoitti, että subjektiivinen terveydentila oli merkittävästi heikentynyt kaikissa kahdeksan aloilla lyhyen aikavälin terveys survey, jossa näkyvimmät erot, verrattuna ikä-ja sukupuoli – sovitettu valvontaa, jotka liittyvät aloilla yleistä terveyttä, elinvoimaa, ja rooli rajoitukset johtuvat emotionaalisia ongelmia. Hospital Anxiety and Depression Scale (HADS) – kyselyssä havaittiin myös kohonneita ahdistuneisuuspisteitä (41). Nämä havainnot huomioon ottaen nccah: ta sairastavilla naisilla olisi harkittava psykologisia parametreja hoidon ohjaamiseksi, ja potilaslähtöisen lähestymistavan yhteydessä on tarjottava psykologista diagnoosia ja tukea.
johtopäätökset
NCCAH: ta pidetään CAH: n yleisempänä ja lievempänä muotona 20-50% entsyymiaktiivisuuden retention vuoksi. Useimmat potilaat voivat hakeutua lääkäriin missä tahansa elämänsä vaiheessa androgeeniylimäärään liittyvien oireiden vuoksi. Näitä ovat ennenaikainen pubarche, nopea kasvu, hirsutismi, akne, kuukautiskierron väärinkäytöksiä, tai hedelmättömyys, sekä monia muita vähemmän näkyviä ilmenemismuotoja. Varhaisen aamun lähtöarvoksi suositellaan 17 OHP: tä hyvänä alustavana seulontatestinä ja LISÄARVIONA ACTH-stimulaatiolla sekä rajatarkastustulosten osalta geneettistä testausta. Yleensä seurannan aikana tulee käyttää androstenedionia eikä 17 OHP-tasoa.
hoitopäätöksen tulee perustua tosiseikkojen arviointiin ja noudattaa yksilöllistä lähestymistapaa. Hoitoa ei aina ole tarkoitettu, ellei potilas ole oireenmukainen, esim.lapset, joilla häpy-ja vartalokarvojen varhainen ja nopea eteneminen, nopea kasvu ja/tai luuston eteneminen, tai naiset, joilla on oligomenorrea, akne, hirsutismi, hedelmättömyys tai näiden ja muiden edellä mainittujen oireiden yhdistelmä. Geneettinen neuvonta on erittäin suositeltavaa nccah naiset, jotka haluavat tulla raskaaksi, sekä genotyypin isä. Potilaan kokonaishoitoon kuuluu lisäksi glukokortikoidihoidon todennäköisten komplikaatioiden tai taudin metaboliaan liittyvien oireiden hoito. Lopuksi, koska monia näiden potilaiden asianmukaiseen hoitoon liittyviä terapeuttisia kysymyksiä ei ole vielä selvitetty, on erittäin tärkeää, että hoitava lääkäri pysyy ajan tasalla kaikesta tämän alan kehityksestä ja sisällyttää uudet tiedot kliiniseen käytäntöönsä. Varmasti, lisää kliinisiä tutkimuksia tällä alalla ovat välttämättömiä.
yhteenvetona voidaan todeta, että nccah-naiselle ihanteellinen lähestymistapa on räätälöity siten, että hänen hoitonsa siirtyy sujuvasti, kun hänet siirretään lapsipotilaasta aikuiseksi endokrinologiksi, sekä oireisiin keskittyvä hoito, joka seuraa häntä koko hänen elämänsä ajan. Koska hyperandrogeeniseen järjestelmään vaikuttavat monet tekijät, on suositeltavaa kannustaa potilaita omaksumaan terveellisempiä elämäntapoja parantamalla ruokailutottumuksiaan, lisäämällä liikuntaa ja pyrkimällä painonpudotukseen. Lisäksi tietyissä tapauksissa psykologinen tuki on usein hyödyllistä. Lisäksi tämän häiriön hoitoon osallistuvien alojen asiantuntijat, kuten ihotautilääkärit, gynekologit ja psykologit, tarvitsevat syvällistä ymmärrystä epäilyttävien tai jo diagnosoitujen nccah-tapausten hallinnasta. Lopuksi muistakaamme amerikkalaisen tähtitieteilijän Vera Cooper Rubinin oivaltava lainaus: ”tiede edistyy parhaiten, kun havainnot pakottavat meidät muuttamaan ennakkokäsityksiämme.”
Tekijäosuudet
kaikki luetellut tekijät ovat antaneet merkittävän, suoran ja älyllisen panoksen teokseen ja hyväksyneet sen julkaistavaksi.
Eturistiriitalausunto
kirjoittajat toteavat, että tutkimus tehtiin ilman kaupallisia tai taloudellisia suhteita, joita voitaisiin pitää mahdollisena eturistiriitana.
1. Speiser PW, valkoinen PC. Synnynnäinen lisämunuaisen hyperplasia. N Engl J Med. (2003) 349:776–88. doi: 10.1056/NEJMra021561
PubMed Abstract | CrossRef Full Text | Google Scholar
2. Turcu AF, Auchus RJ. Adrenal steroidogenesis and congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. (2015) 44:275–96. doi: 10.1016/j.ecl.2015.02.002
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Speiser PW. Nonclassic adrenal hyperplasia. Rev Endocr Metab Disord. (2009) 10:77–82. doi: 10.1007/s11154-008-9097-x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Gidlöf S, Farhammar H, Thilén A, von Döbeln U, Ritzén M, Wedell A, et al. Sata vuotta synnynnäistä lisämunuaisen hyperplasiaa Ruotsissa: retrospektiivinen, väestöpohjainen kohorttitutkimus. Lancet Diabetes Endocrinol. (2013) 1:35–42. doi: 10.1016/S2213-8587(13)70007-X
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, New MI. Korkea taajuus ei-Luokka steroidi 21-hydroksylaasin puutos. Olen J. Hum Genet. (1985) 37:650–67.
PubMed Abstract | Google Scholar
6. Wilson RC, Nimkarn S, Dumic M, Obeid J, Azar MR, Azar M, et al. Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Mol Genet Metab. (2007) 90:414–21. doi: 10.1016/j.ymgme.2006.12.005
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Miller WL. Clinical review 54: genetics, diagnosis, and management of 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1994) 78:241–6. doi: 10.1210/jcem.78.2.8106606
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Witchel SF, Azziz R. Nonclassic congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2010) 2010:625105. doi: 10.1186/1687-9856-2010-625105
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Wilson RC, Mercado AB, Cheng KC, New MI. Steroidi 21-hydroksylaasin puutos: genotyyppi ei välttämättä ennusta fenotyyppiä. J Clin Endocrinol Metab. (1995) 80:2322–9. doi: 10.1210 / jc.80.8.2322
CrossRef Full Text/Google Scholar
10. Valkoinen PC. Vastasyntyneen seulonta synnynnäisen lisämunuaisen liikakasvun varalta. Nat Rev Endocrinol. (2009) 5:490–8. doi: 10.1038 / norendo.2009.148
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Kohn B, Levine LS, Pollack MS, Pang S, Lorenzen F, Levy D, et al. Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. (1982) 55:817–27. doi: 10.1210/jcem-55-5-817
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Moran C, Azziz R, Carmina E, Dewailly D, Fruzzetti F, Ibañez L, et al. 21-hydroksylaasin puutteellinen ei-klassinen lisämunuaisen hyperplasia on etenevä häiriö: monikeskustutkimus. Am J Synnytyslääkäri Gynecol. (2000) 183:1468–74. doi: 10.1067 / mob.2000.108020
PubMed Abstract | CrossRef Full Text/Google Scholar
13. Azziz R, Hincapie LA, Knochenhauer ES, Dewailly D, Fox L, Boots LR. Screening for 21-hydroxylase-deficient nonclassic lisämunuaisen hyperplasia among hyperandrogenic women: a prospective study. Fertil Steril. (1999) 72:915–25. doi: 10.1016/S0015-0282(99)00383-0
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Livadas S, Dracopoulou M, Dastamani A, Sertedaki A, Maniati-Christidi M, Magiakou A-M, et al. Kliinisten, hormonaalisten ja molekulaaristen löydösten kirjo 280 henkilöllä, joilla on cyp21a2-geenin mutaatioiden aiheuttama ei-luokkainen synnynnäinen lisämunuaisen hyperplasia. Clin Endocrinol (Oxf). (2015) 82:543–9. doi: 10.1111 / cen.12543
PubMed Abstract | CrossRef Full Text/Google Scholar
15. Bidet M, Bellanné-Chantelot C, Galand-Portier M-B, Tardy V, Billaud L, Laborde K, et al. Kliininen ja molekyylitason karakterisointi kohortista, jossa oli 161 sukukypsää naista, joilla oli 21-hydroksylaasin puutoksesta johtuva synnynnäinen lisämunuaisen hyperplasia ja 330 perheenjäsentä. J Clin Endocrinol Metab. (2009) 94:1570–8. doi: 10.1210 / jc.2008-1582
PubMed Abstract | CrossRef Full Text/Google Scholar
16. Escobar-Morreale HF, Sanchón R, San Millán JL. Prospektiivinen tutkimus ei-klassisesta synnynnäisestä lisämunuaisen hyperplasiasta naisilla, joilla on hyperandrogeenisia oireita ja merkkejä. J Clin Endocrinol Metab. (2008) 93:527–33. doi: 10.1210 / jc.2007-2053
PubMed Abstract | CrossRef Full Text/Google Scholar
17. Dewailly D, Vantyghem-Haudiquet MC, Sainsard C, Buvat J, Cappoen JP, Ardaens K, et al. Kliiniset ja biologiset fenotyypit myöhäisessä 21-hydroksylaasin puutoksessa. J Clin Endocrinol Metab. (1986) 63:418–23. doi: 10.1210 / jcem-63-2-418
PubMed Abstract | CrossRef Full Text/Google Scholar
18. Gönç EN, Ozön ZA, Alikaşifoglu A, Engiz O, Bulum B, Kandemir N. (2011). Onko basal seerumin 17-OH progesteroni luotettava parametri ennustaa ei-Luokka synnynnäinen lisämunuaisen hyperplasia ennenaikainen adrenarche? Turk J Pediatr. 53:274–80.
PubMed Abstract / Google Scholar
19. Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, et al. Synnynnäinen lisämunuaisen hyperplasia johtuu steroidi 21-hydroksylaasin puutos: hormonitoimintaa society kliinisen käytännön ohje. J Clin Endocrinol Metab. (2010) 95:4133–60. doi: 10.1210 / jc.2009-2631
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Dacou-Voutetakis C, Dracopoulou M. High incidence of molecular defects of the CYP21 gene in patients with premature adrenarche. J Clin Endocrinol Metab. (1999) 84:1570–4. doi: 10.1210/jc.84.5.1570
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Ambroziak U, Kepczynska-Events I, Kurylowicz, Małunowicz EM, Wójcicka, Miskiewicz P, et al. Diagnoosi ei-klassisesta synnynnäisestä lisämunuaiskuoren hyperplasiasta, joka johtuu 21-hydroksylaasin puutteesta, joka perustuu seerumin basal-tai post-ACTH-stimulaatioon 17-hydroksiprogesteroni, voi johtaa väärään positiiviseen diagnoosiin. Clin Endocrinol (Oxf). (2016) 84:23–9. doi: 10.1111 / price 12935
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Ambroziak Y, Kepczynska-Events I, Kurylowicz, Wysłouch-Cieszyńska A, Małunowicz EM, Bartoszewicz, et al. LC-MS / MS parantaa seulontaa kohti 21-hydroksylaasin puutosta. Gynecol Endokrinol. (2015) 31:296–300. doi: 10.3109/09513590.2014.994599
PubMed Abstract | CrossRef Full Text | Google Scholar
23. bbc, CC, Goldman MM, Zhang K, Clark-n, Reitz RE Welt CK. Kolmentoista steroidihormonin samanaikainen mittaus naisilla, joilla on munasarjojen monirakkulatauti ja kontrolli naisilla nestekromatografian-tandem-massaspektrometrian avulla. PLoS Yksi. (2014) 9:e93805. doi: 10.1371 / lehti.pone.0093805
PubMed Abstract | CrossRef Full Text/Google Scholar
24. Ray ja, Kushnir MM, Yost RA, Rockwood AL, Wayne Meikle A. suorituskyvyn parantaminen 5 endogeenisen steroidin mittauksessa LC-MS / MS yhdistettynä differentiaali-ioniliikkuvuusspektrometriaan. Clin Chim Acta. (2015) 438:330–6. doi: 10.1016 / J.cca.2014.07.036
PubMed Abstract | CrossRef Full Text/Google Scholar
25. Stoupa A, González-Briceño L,Pinto G, Samara-Boustani D, Thalassinos C, Flechtner I, et al. Riittämätön kortisolin vaste tetrakosaktidin (Synakthen®) testiin ei-klassisessa synnynnäisessä lisämunuaisen hyperplasiassa: poikkeus sääntöön? Horm Res Paedatr. (2015) 83:262–7. doi: 10.1159/000369901
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. (1992) 90:584–95. doi: 10.1172/JCI115897
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Temeck JW, Pang SY, Nelson C, Uusi MI. Steroidogeneesin geenivirheet ennenaikaisessa pubarchessa. J Clin Endocrinol Metab. (1987) 64:609–17. doi: 10.1210 / jcem-64-3-609
PubMed Abstract | CrossRef Full Text/Google Scholar
28. Voutilainen R, Jääskeläinen J. Premature adrenarche: etiologia, kliiniset löydökset ja seuraukset. J Steroidi Biochem Mol Biol. (2015) 145:226–36. doi: 10.1016 / j.jsbmb.2014.06.004
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Oberfield SE, Sopher AB, Gerken AT. Approach to the girl with early onset of pubic hair. J Clin Endocrinol Metab. (2011) 96:1610–22. doi: 10.1210/jc.2011-0225
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Janus D, Wojcik M, Malunowicz E, Starzyk JB. Toistuva labiaalinen kiinnike 15 kuukauden ikäisellä lapsella, jolla on oireeton ei-klassinen synnynnäinen lisämunuaisen hyperplasia, joka johtuu 21-hydroksylaasin puutteesta. J Pediatr Endocrinol Metab. (2012) 25:1017–21. doi: 10.1515/jpem-2012-0190
PubMed Abstract | CrossRef Full Text | Google Scholar
31. Gopal-Kothandapani JS, Petkar, O ’ Shea E, Banerjee I. Perianal hiukset epätavallinen esitys ei-klassinen synnynnäinen lisämunuaisen hyperplasia. BMJ Case Rep. (2013) 2013:bcr2013010123. doi: 10.1136/bcr-2013-010123
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Falhammar H, Nordenström A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine. (2015) 50:32–50. doi: 10.1007/s12020-015-0656-0
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Chrousos GP, Loriaux DL, Mann DL, Cutler GB. Late-onset 21-hydroxylase deficiency mimicking idiopathic hirsutism or polycystic ovarian disease. Ann Intern Med. (1982) 96:143–8. doi: 10.7326/0003-4819-96-2-143
CrossRef Full Text | Google Scholar
34. Witchel SF. Non-classic congenital adrenal hyperplasia. Steroids. (2013) 78:747–50. doi: 10.1016/j.steroids.2013.04.010
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Carmina E, Rosato F, Jannì A, Rizzo M, Longo RA. Extensive clinical experience: relative prevalence of different androgen excess disorders in 950 women referred because of clinical hyperandrogenism. J Clin Endocrinol Metab. (2006) 91:2–6. doi: 10.1210/jc.2005-1457
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Moran C, Azziz R, Weintrob N, Witchel SF, Rohmer V, Dewailly D, et al. Lisääntymistulos naisilla, joilla on 21-hydroksylaasi-puutteellinen ei-klassinen lisämunuaisen hyperplasia. J Clin Endocrinol Metab. (2006) 91:3451–6. doi: 10.1210 / jc.2006-0062
PubMed Abstract | CrossRef Full Text/Google Scholar
37. Levin JH, Carmina E, Lobo RA. Onko sopimaton gonadotropiinin eritystä potilailla, joilla on munasarjojen monirakkulatauti samanlainen kuin potilailla, joilla on aikuisiän synnynnäinen lisämunuaisen hyperplasia? Fertil Steril. (1991) 56:635–40. doi: 10.1016/S0015-0282(16)54592-0
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Carmina E, Lobo RA. Ovarian suppression reduces clinical and endocrine expression of late-onset congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Fertil Steril. (1994) 62:738–43. doi: 10.1016/S0015-0282(16)56998-2
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Ludwig E. luokittelu androgeneettisen hiustenlähtö (yleinen kaljuuntuminen) esiintyy naispuoliseen sukupuoleen. BRJ Dermatol. (1977) 97:247–54. doi: 10.1111 / j. 1365-2133. 1977.tb15179.x
PubMed Abstract | CrossRef Full Text | Google Scholar
40. Trakakis E, Papadavid E, Dalamaga M, Koumaki D, Stavrianeas N, Rigopoulos D, et al. 21-hydroksylaasin puutoksesta johtuvan ei-klassisen synnynnäisen lisämunuaisen hyperplasian Prevalence in Greek women with akne: a hospital-based cross-sectional study. J Eur Acad Dermatol Venereol. (2013) 27:1448–51. doi: 10.1111 / j.1468-3083.2012. 04613.x
PubMed Abstract | CrossRef Full Text | Google Scholar
41. Arlt W, Willis DS, Wild SH, Krone N, Doherty EJ, Hahner s, et al. Synnynnäisestä lisämunuaisen hyperplasiasta kärsivien aikuisten terveydentila: kohorttitutkimus, johon osallistui 203 potilasta. J Clin Endocrinol Metab. (2010) 95:5110–21. doi: 10.1210 / jc.2010-0917
PubMed Abstract | CrossRef Full Text/Google Scholar
42. Saygili F, Oge A, Yilmaz C. Hyperinsulinemia ja insuliiniherkkyys naisilla, joilla on 21-hydroksylaasin puutoksesta johtuva ei-luokkainen synnynnäinen lisämunuaisen hyperplasia: seerumin leptiinipitoisuuden ja kroonisen hyperinsulinemian välinen suhde. Horm Res. (2005) 63: 270-4. doi: 10.1159/000086363
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Speiser PW, Serrat J, New MI, Gertner JM. Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1992) 75:1421–4. doi: 10.1210/jc.75.6.1421
PubMed Abstract | CrossRef Full Text | Google Scholar
44. Han TS, Stimson RH, Rees DA, Krone N, Willis DS, Conway GS, et al. Glukokortikoidihoito ja terveysvaikutukset aikuisilla, joilla on synnynnäinen lisämunuaisen hyperplasia. Clin Endocrinol. (2013) 78:197–203. doi: 10.1111 / cen.12045
PubMed Abstract | CrossRef Full Text/Google Scholar
45. Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. suun kautta otettavien kortikosteroidien käyttö ja murtumariski. J Bone Miner Res. (2000) 15: 993-1000. doi: 10.1359 / jbmr.2000.15.6.993
PubMed Abstract | CrossRef Full Text | Google Scholar
46. Nebesio TD, Renbarger JL, Nabhan ZM, Ross SE, Slaven JE, Li L, et al. Differential effects of hydrocortisone, prednisone, and dexamethasone on hormonal and pharmacokinetic profiles: a pilot study in children with congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2016) 2016:17. doi: 10.1186 / s13633-016-0035-5
PubMed Abstract | CrossRef Full Text/Google Scholar
47. Weintrob n, Dickerman Z, Sprecher E, Galatzer a, Pertzelan A. Ei-klassinen 21-hydroksylaasin puutos lapsenkengissä ja lapsuudessa: hoidon aloittamisajan vaikutus murrosikään ja lopulliseen pituuteen. Eur J Endocrinol. (1997) 136:188–95. doi: 10.1530 / eje.0.1360188
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Bonfig W, Bechtold S, Schmidt H, Knorr D, Schwarz HP. Reduced final height outcome in congenital adrenal hyperplasia under prednisone treatment: deceleration of growth velocity during puberty. J Clin Endocrinol Metab. (2007) 92:1635–9. doi: 10.1210/jc.2006-2109
PubMed Abstract | CrossRef Full Text | Google Scholar
49. Dörr HG. Kasvu potilailla, joilla on klassinen synnynnäinen lisämunuaisen hyperplasia, joka johtuu 21-hydroksylaasin puutteesta. Horm Res. (2007) 68(Suppl 5): 93-9. doi: 10.1159/000110587
PubMed Abstract | CrossRef Full Text | Google Scholar
50. Hoepffner W, Kaufhold A, Willgerodt H, Keller E. potilaat, joilla on klassinen synnynnäinen lisämunuaisen hyperplasia, joka johtuu 21-hydroksylaasin puutteesta, voivat saavuttaa tavoitekorkeuden: Leipzigin kokemus. Horm Res. (2008) 70:42-50. doi: 10.1159/000129677
PubMed Abstract | CrossRef Full Text | Google Scholar
51. van der Kaay D, van den Akker E. Ultralow-dose dexamethasone to preserve endogenous cortisol stress response in nonclassical congenital adrenal hyperplasia: a new promising treatment. Int J Endocrinol Metab. (2014) 12:e14657. doi: 10.5812/ijem.14657
CrossRef Full Text | Google Scholar
52. Lin-Su K, Vogiatzi MG, Marshall I, Harbison MD, Macapagal MC, Betensky B, et al. Treatment with growth hormone and luteinizing hormone releasing hormone analog improves final adult height in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. (2005) 90:3318–25. doi: 10.1210/jc.2004-2128
PubMed Abstract | CrossRef Full Text | Google Scholar
53. McCann-Crosby B, Chen M-J, Lyons SK, Lin Y, Axelrad M, Dietrich JE, et al. Nonclassical congenital adrenal hyperplasia: hoito-ja siirtymäkohteet. Pediatr Endocrinol Rev. (2014) 12:224-38.
PubMed Abstract/Google Scholar
54. Ogilvie CM, Crouch NS, Rumsby G, Creighton SM, Liao L-M, Conway GS. Synnynnäinen lisämunuaisen hyperplasia aikuisilla: lääketieteellisten, kirurgisten ja psykologisten kysymysten tarkastelu. Clin Endocrinol. (2006) 64:2–11. doi: 10.1111 / j.1365-2265.2005. 02410.x
PubMed Abstract | CrossRef Full Text | Google Scholar
55. Mah PM, Jenkins RC, Rostami-Hodjegan A, Newell-Price J, Doane A, Ibbotson V, et al. Weight-related dosing, timing and monitoring hydrocortisone replacement therapy in patients with adrenal insufficiency. Clin Endocrinol. (2004) 61:367–75. doi: 10.1111/j.1365-2265.2004.02106.x
PubMed Abstract | CrossRef Full Text | Google Scholar
56. Diaz A, Laufer MR Breech LLAA of PC on a, Care AC of O and GC on AH. Kuukautiset tytöillä ja nuorilla: kuukautiskierron käyttäminen elintärkeänä merkkinä. Lastentautioppi. (2006) 118:2245–50. doi: 10.1542 / peds.2006-2481
CrossRef Full Text/Google Scholar
57. Merke DP, Poppas DP. Synnynnäisestä lisämunuaisen hyperplasiasta kärsivien nuorten hoito. Lancet Diabetes Endocrinol. (2013) 1:341–52. doi: 10.1016/S2213-8587(13)70138-4
PubMed Abstract | CrossRef Full Text | Google Scholar
58. Verma S, Green-Golan L, VanRyzin C, Drinkard B, Mehta SP, Weise M, et al. Adrenomedullary function in patients with nonclassic congenital adrenal hyperplasia. Horm Metab Res. (2010) 42:607–12. doi: 10.1055/s-0030-1253385
PubMed Abstract | CrossRef Full Text | Google Scholar
59. New MI. Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2006) 91:4205–14. doi: 10.1210/jc.2006-1645
PubMed Abstract | CrossRef Full Text | Google Scholar
60. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2018) 103:4043–88. doi: 10.1210/jc.2018-01865
PubMed Abstract | CrossRef Full Text | Google Scholar
61. Kruse B, Riepe FG, Krone N, Bosinski HAG, Kloehn S, Partsch CJ, et al. Congenital adrenal hyperplasia – how to improve the transition from adolescence to adult life. Exp Clin Endocrinol Diabetes. (2004) 112:343–55. doi: 10.1055/s-2004-821013
PubMed Abstract | CrossRef Full Text | Google Scholar
62. Gastaud F, Bouvattier C, Duranteau L, Brauner R, Thibaud E, kuten F, et al. Heikentyneet seksuaaliset ja lisääntymistulokset naisilla, joilla on synnynnäinen lisämunuaisen hyperplasia. J Clin Endocrinol Metab. (2007) 92:1391–6. doi: 10.1210 / jc.2006-1757
PubMed Abstract | CrossRef Full Text/Google Scholar
63. Mercè Fernández-Balsells M, Muthusamy K, Smushkin G, Lampropulos JF, Elamin MB, Abu Elnour NO, et al. Prenatal deksametasone use for the prevention of virilization in pregnants at risk for classical congenitional lisämunuaisen hyperplasia because of 21-hydroxylase (CYP21A2) deficiency: a systematic review and meta-analyses. Clin Endocrinol. (2010) 73:436–44. doi: 10.1111 / j.1365-2265.2010. 03826.x
CrossRef Full Text | Google Scholar
64. Bidet M, Bellanné-Chantelot C, Galand-Portier MB, Golmard JL, Tardy V, Morel Y, et al. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2010) 95:1182–90. doi: 10.1210/jc.2009-1383
PubMed Abstract | CrossRef Full Text | Google Scholar
65. Birnbaum MD, Rose LI. Late onset adrenocortical hydroxylase deficiencies associated with menstrual dysfunction. Obstet Gynecol. (1984) 63:445–51.
PubMed Abstract | Google Scholar
66. Cuhaci N, Aydin C, Yesilyurt A, Pinarli FA, Ersoy R, Cakir B. Nonclassical congenital adrenal hyperplasia and pregnancy. Case Rep Endocrinol. (2015) 2015:296924. doi: 10.1155/2015/296924
PubMed Abstract | CrossRef Full Text | Google Scholar
67. Deneux C, Tardy V, Anne D, Mornet E, Billaud L, Charron D, et al. Fenotyypin ja genotyypin korrelaatio 56 naisella, joilla oli ei-Luokka synnynnäinen lisämunuaisen hyperplasia, joka johtui 21-hydroksylaasin puutteesta. J Clin Endocrinol Metab. (2001) 86:207–13. doi: 10.1210 / jcem.86.1.7131
PubMed Abstract | CrossRef Full Text/Google Scholar
68. Simonetti L, Bruque CD, Fernández CS, Benavides-Mori B, Delea M, Kolomenski je, et al. CYP21A2 mutation update: Comprehensive analysis of databases and published genetic variants. Hum Mutat. (2018) 39:5–22. doi: 10.1002 / humu.23351
PubMed Abstract | CrossRef Full Text | Google Scholar
69. OMIM Entry – * 613815 – CYTOCHROME P450 FAMILY 21 SUBFAMILY A POLYPEPTIDE 2; CYP21A2.
Google Scholar
70. Miller WL, Witchel SF. Prenatal treatment of congenital adrenal hyperplasia: risks outweigh benefits. Am J Obstet Gynecol. (2013) 208:354–9. doi: 10.1016/j.ajog.2012.10.885
PubMed Abstract | CrossRef Full Text | Google Scholar
71. Lekarev O, New MI. Adrenal disease in pregnancy. Best Pract Res Clin Endocrinol Metab. (2011) 25:959–73. doi: 10.1016/j.beem.2011.08.004
PubMed Abstract | CrossRef Full Text | Google Scholar
72. New MI, Abraham M, Yuen T, Lekarev O. An update on prenatal diagnosis and treatment of congenital adrenal hyperplasia. Semin Reprod Med. (2012) 30:396–9. doi: 10.1055/s-0032-1324723
PubMed Abstract | CrossRef Full Text | Google Scholar
73. Kazmi D, Bailey J, Yau M, Abu-Amer W, Kumar A, Low M, et al. New developments in prenatal diagnosis of congenital adrenal hyperplasia. J Steroid Biochem Mol Biol. (2017) 165:121–3. doi: 10.1016/j.jsbmb.2016.06.016
PubMed Abstract | CrossRef Full Text | Google Scholar
74. Clayton PE, Miller WL, Oberfield SE, Ritzén EM, Sippell WG, Speiser PW ym. Consensus statement on 21-hydroxylase deficience from the European Society for Pediatric Endocrinology and the lawson wilkins pediatric endocrine society. Horm Res. (2002) 58: 188-95. doi: 10.1159/000065490
PubMed Abstract | CrossRef Full Text | Google Scholar
75. Nandagopal R, Sinaii N, Avila NA, Van Ryzin C, Chen W, Finkielstain GP, et al. Fenotyyppinen profilointi vanhemmista, joilla on kryptinen ei-klassinen synnynnäinen lisämunuaisen hyperplasia: löydökset 145: ssä toisiinsa liittymättömässä perheessä. Eur J Endocrinol. (2011) 164:977–84. doi: 10.1530/EJE-11-0019
PubMed Abstract | CrossRef Full Text | Google Scholar
76. Takasu N, Nakachi K, Higa H. Gravesin kilpirauhasen liikatoiminnan kehittyminen aiheutti lisämunuaiskriisin potilaalla, jolla oli aiemmin tunnistamaton ei-klassinen 21-hydroksylaasin puutos. Harjoittelija. (2010) 49:1395–400. doi: 10.2169 / Internal Medicine.49.3573
PubMed Abstract | CrossRef Full Text | Google Scholar
77. Meyer-Bahlburg HFL, Dolezal C, Baker SW, Carlson AD, Obeid JS, New MI. Prenatal androgenization affects gender-related behavior but not gender identity in 5-12-year-old girls with congenital adrenal hyperplasia. Arch Sex Behav. (2004) 33:97–104. doi: 10.1023/B:ASEB.0000014324.25718.51
CrossRef Full Text | Google Scholar
78. Meyer-Bahlburg HFL, Dolezal C, Baker SW, New MI. Seksuaalinen suuntautuminen naisilla, joilla on klassinen tai ei-klassinen synnynnäinen lisämunuaisen hyperplasia raskauden androgeeniylimäärän funktiona. Arch Sex Behaven. (2008) 37:85–99. doi: 10.1007 / s10508-007-9265-1
PubMed Abstract / CrossRef Full Text / Google Scholar