Modrá bělma je způsobeno tím, že hubenost a transparentnost kolagenových vláken, skléry, které umožňují vizualizaci hlubších uvea (Obrázek 1). Skléry jsou bílé vnější plášť oka, kolem iris může být ředěn ve vrozené onemocnění, jako je osteogenesis imperfecta 1). Další nemoci spojené s modré skléry obsahuje více pojivové tkáně, jako jsou křehké rohovky syndrom 2), Marshall a Stickler syndrom 3), BÁSNĚ (polyneuropatie, organomegaly, endocrinopathy, monoklonální protein a kožní změny) syndrom 4), Marfanův syndrom, Ehlers–Danlosův syndrom, pseudoxanthoma elasticum, a Willems De Vries syndrom, abychom jmenovali alespoň některé. Poruchy kostí a krve také na seznamu zahrnují anémii Diamond–Blackfan, těžkou anémii s nedostatkem železa, juvenilní Pagetovu chorobu a nedostatek kyselé fosfatázy 5).
těžké formy osteogenesis imperfecta jsou nejčastěji diagnostikovány na počátku života, ale mírné případy mohou být zaznamenány až později v životě. Modrošedá barva skléry je způsobena podkladovými choroidálními žilkami, které se projevují. To je způsobeno tím, že sklera je tenčí než obvykle, protože defektní kolagen typu I se netvoří správně 6). Sclera je hustá, špatně prokrvené pojivové tkáně struktury složené typy I, III, IV, V, VI a VIII kolagen, stejně jako elastin, proteoglykany a glykoproteiny 7).
ve Spojených státech se výskyt osteogenesis imperfecta odhaduje na jeden na 20 000 živě narozených dětí. Odhaduje se, že ve Spojených státech je osteogenesis imperfecta postiženo 20 000 až 50 000 lidí 8).
Lidé s osteogenesis imperfecta se rodí s defektní pojivovou tkání, nebo bez schopnosti ji vytvořit, obvykle kvůli nedostatku kolagenu Typu I. Tento nedostatek vzniká substitucí aminokyselin glycinu na objemnější aminokyseliny ve struktuře kolagenové trojité šroubovice. V důsledku toho může tělo reagovat hydrolyzací nesprávné struktury kolagenu 9). Pokud tělo nemá zničit nesprávné kolagenu; vztah mezi vlákna kolagenu a hydroxyapatitu krystaly tvoří kosti se mění, což způsobuje křehkost. Jako genetická porucha byla osteogenesis imperfecta historicky považována za autozomálně dominantní poruchu kolagenu typu I. Většina případů byla způsobena mutacemi v genech COL1A1 a COL1A2 10). V posledních několika letech došlo k identifikaci autosomálně recesivních forem. Bylo popsáno nejméně sedm podmnožin, i když čtyři hlavní podtypy jsou nejčastější a pohybují se od mírných po těžké. Jedinci s osteogenesis imperfecta typu I mají malou kostní deformitu, přetrvávající modrou skléru, téměř normální výšku do dospělosti a >50% pravděpodobnost ztráty sluchu do dospělosti. Pacienti s perinatální letální (typ II) osteogenesis imperfecta vykazují největší závažnost s mnohočetnými zlomeninami in utero nebo od porodu. Tito pacienti jsou obvykle mrtvě narození nebo umírají brzy. Závažnost osteogenesis imperfecta závisí na specifickém defektu genu. Většina případů osteogenesis imperfecta je zděděna od rodiče. Některé případy jsou však výsledkem nových genetických mutací. Osoba s osteogenesis imperfecta má 50% šanci předat gen a nemoc svým dětem 11).
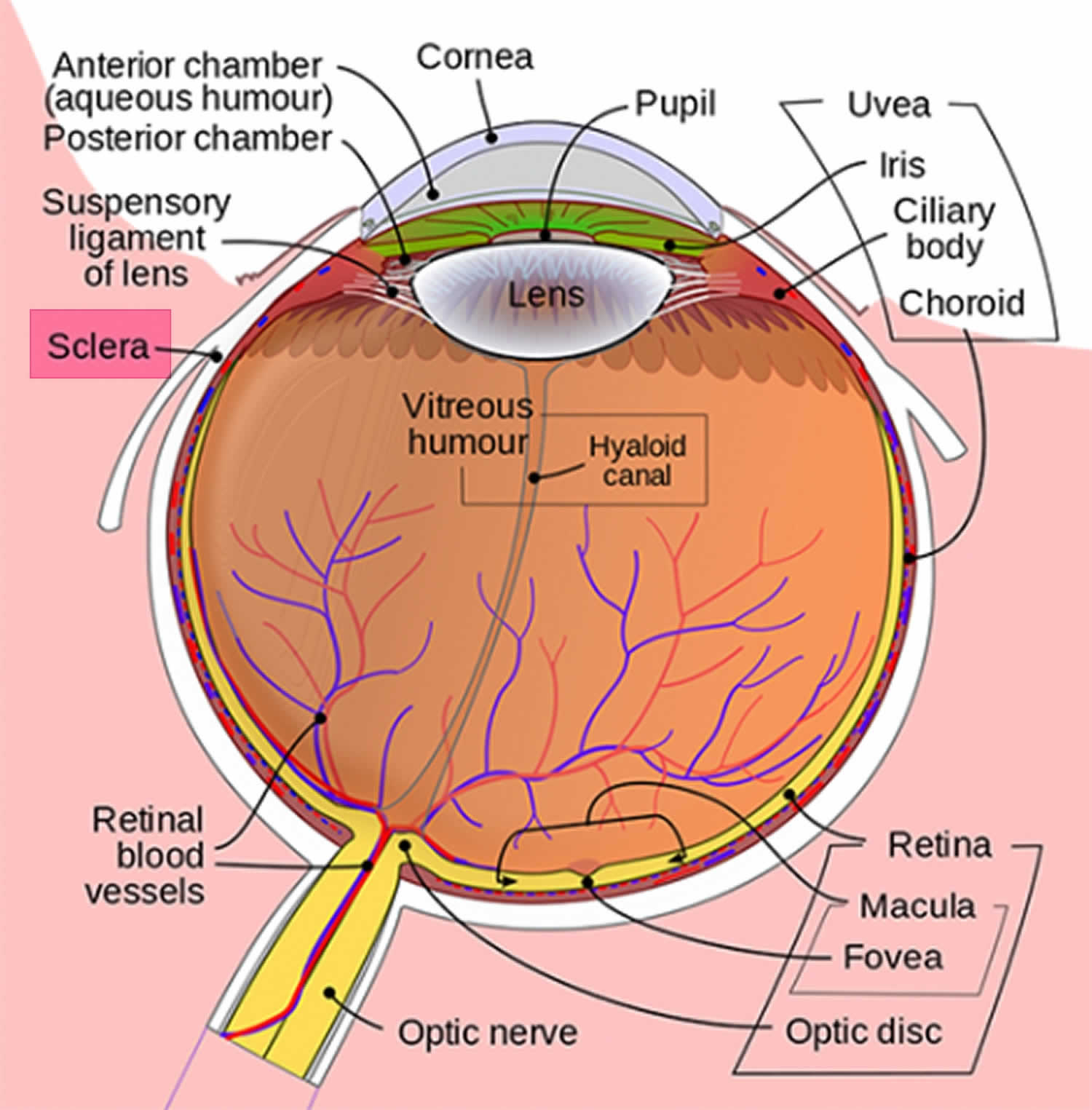
Obrázek 1. Anatomie oka
Obrázek 2. Modré skléry
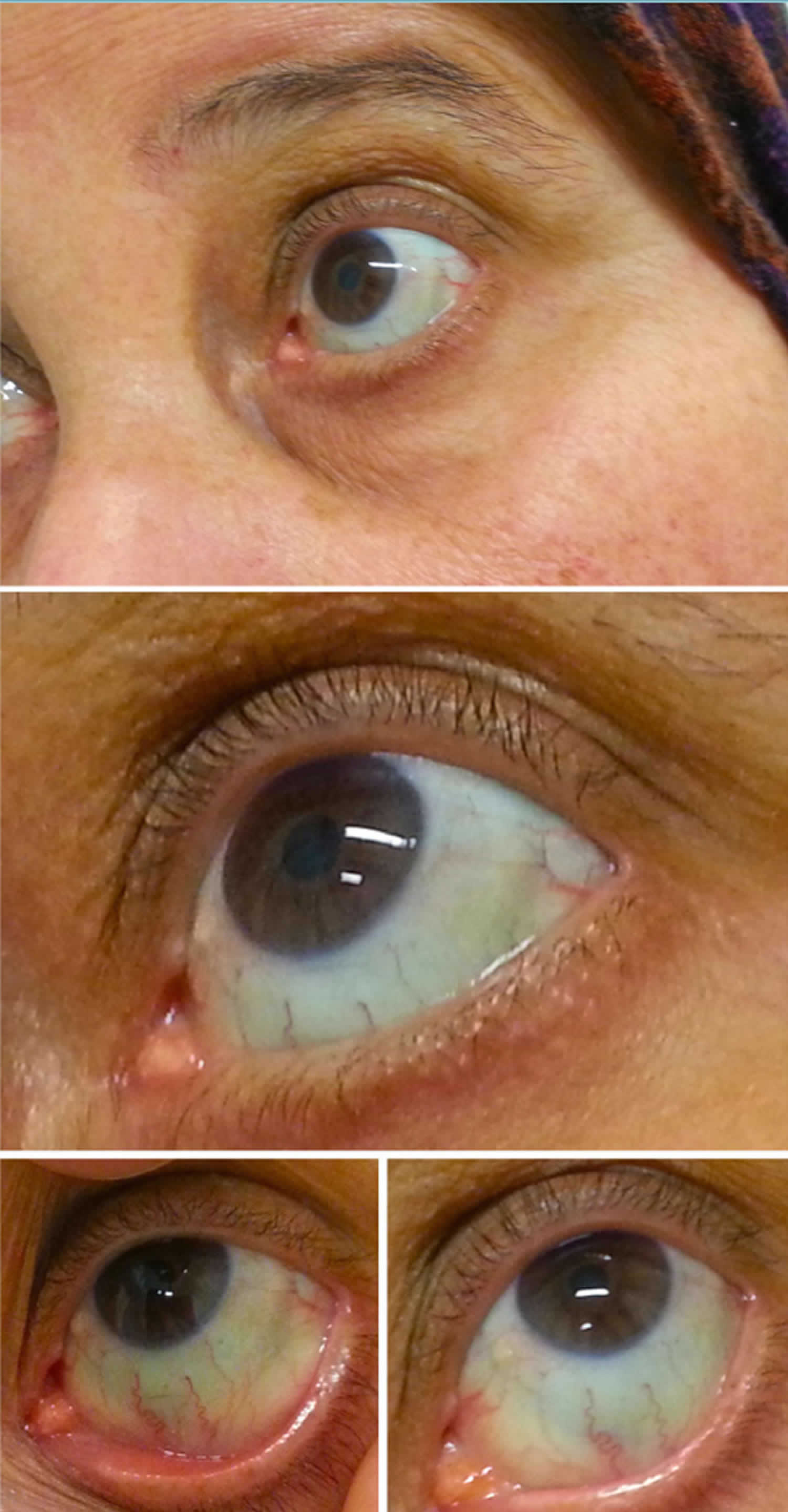
Modré skléry příčiny
Modré skléry je nejvíce konzistentní projev osteogenesis imperfecta, která je důsledkem mutací v COLIA1 a COL1A2, kódující prokolagenu typu I. Klasifikace tohoto stavu (typy IV-VI) však byly identifikovány s normálními sklerami. Osteogenesis imperfecta je také spojena s abnormální křehkostí kostí a hluchotou.
Křehké rohovky, modré skléry, a červené vlasy jsou spojeny s křehké rohovky syndrom, onemocnění, které také představuje s kostní, zubní a kožní abnormality, 13). Bylo zjištěno, že mutace missense v ZNF469 je příčinou onemocnění.
Jiných hlubších stavů spojených s tvorbou modré skléry patří Ehlers-Danlos syndrom (typ VI), pseudoxanthoma elasticum, rohovky plana, periferní sclerocornea, buphthalmos, keratokonus, keratoglobus, vysoká myopie, ciliární/rovníková staphyloma, oculodermal melanocytosis a microcornea. Zřídka, modré skléry se vyskytuje s Hallermann-Streiff syndrom, Marfanův syndrom, Turnerův syndrom, Cheney syndrom, Menkes syndrom, pyknodysostosis, křehké rohovky, nebo ektodermální dysplazie.
Modré skléry, může dojít také v normální kojence během prvních několika měsíců života; nicméně, vytrvalost modré zabarvení v průběhu času může naznačovat přítomnost zvýšeného nitroočního tlaku. Předčasně narozené děti často vykazují modré sklerózy, zejména kavkazského původu.
Modrá sklera se může také vyskytovat izolovaně jako dědičná autozomálně dominantní nebo autozomálně recesivní anomálie 14).
Modré skléry příznaky
Modré skléry dárky s modravým vzhled skléry a může být spojena s patologickými nebo jiné patologické příčiny. Mezi další oční charakteristiky poruch pojivové tkáně spojené s modrou sklérou patří tenká rohovka, epikantální záhyb, krátkozrakost, keratokonus a angioidní pruhy. Systémové vlastnosti pojivové tkáně poruchy spojené s modré skléry patří kožní abnormality, srdeční abnormality, kyfoskolióza, joint hypermobility, křehké kosti, sluchu, anomálie, vaskulární abnormality, a gastrointestinální abnormality 15).
Modré skléry diagnóza
Diagnostické vyšetření modré skléry involvesexternal vyšetření, štěrbinové lampě biomikroskopii,a systémové hodnocení pro přidružené poruchy.Ačkoli neexistuje žádný definitivní test na osteogenesis imperfecta, genetictesting může potvrdit nebo vyloučit známé mutace 16).
léčba modré sklery
léčba modré sklery zahrnuje diagnostiku a léčbu základní příčiny. Modrá sklera je většinou způsobena genetickými syndromy a v menší míře u nongenetických poruch a může se objevit jako vedlejší účinek příjmu léků. Modré skléry jsou obvykle spojeny s vrozenou syntézu kolagenu poruchy, jako je osteogenesis imperfecta, Marfanův syndrom, Ehlers–Danlosův syndrom, pseudoxanthoma elasticum, a Willems De Vries syndrom, abychom jmenovali alespoň některé. Poruchy kostí a krve také na seznamu zahrnují anémii Diamond–Blackfan, těžkou anémii s nedostatkem železa, juvenilní Pagetovu chorobu a nedostatek kyselé fosfatázy 17). Získané modré skléry byly popsány u dospělých s oční melanóza, spojivkového melanomu, alkaptonuria a Addisonova choroba. Vhodná doporučení k pediatrickým a ortopedickým hodnocením mohou být indikována u neočních projevů 18).
genetické poradenství pro související dědičné poruchy může být prospěšné pro pacienty s modrou sklerou a dalšími projevy systémového onemocnění.
chirurgický zákrok může být indikován v případech extrémního ztenčení a perforace. Strukturální podpora může být poskytována zachovalou sklerální štěpu orautologous fascia lata, a to zejména v případech vyžadujících šití zařízení, jako jsou trubky implantát 19). Systémová vyšetření a léčba by měla být považována za léčbu základní patologie.
prognóza modré sklery
prognóza se mění s přítomností očních a systémových projevů základní poruchy. Pacienti s modrou sklerou mají zvýšené riziko ruptury glóbu nebo intraoperační perforace sklery během rutinní operace očí. Systémové poruchy jsou také převládající u pacientů, jako je karotid-kavernózní píštěl, ruptura arterií,ztráta sluchu a zlomeniny kostí 20).
Odkazy