- Definice a Prevalence
- Diagnostika
- NCCAH Projevy u Žen
- projevy v dětství
- Projevy v Dospívání a dospělosti
- NCCAH Vedení Z První Projev na Dospělý Život
- Řízení V Dětství
- Řízení Během Dospívání
- Pacientů Léčených Od Dětství
- Samice S Prvním Projevem Během Dospívání nebo Hyperandrogenních Příznaky Po Ukončení Léčby,
- NCCAH a Reprodukce
- Subfertility
- potraty
- plánování plodnosti
- Prenatální Léčba CAH
- Léčba Během Těhotenství
- Další Speciální Otázky
- zvládání Stresu v NCCAH
- Psychobiologické aspekty v NCCAH
- závěry
- Autor Příspěvky
- Prohlášení o střetu zájmů
Definice a Prevalence
Kongenitální adrenální hyperplazie (CAH) zahrnuje rodině autosomálně recesivní onemocnění charakterizované mírnou až akutně zhoršené syntézy kortizolu vzhledem k nedostatku v jednom z pěti nadledvin steroidogenní enzymy potřebné pro produkci kortizolu (1, 2). Konvenčně, CAH je rozdělena do (a) klasické (CCAH), projevující se solí-plýtvání nebo simple virilizing formu, která se projevuje při narození nebo v novorozeneckém období, a (b) non-klasické (NCCAH), což představuje méně závažnou formou onemocnění, které postrádá pohlavní nejednoznačnost, není bezprostředně život ohrožující, a představuje později v životě, nebo zůstává bez příznaků, nebo je chybně diagnostikována jako jiné onemocnění (3). Hranice napříč různými formami CAH však nejsou striktně definovány, což má tendenci zvyšovat výzvy spojené s touto poruchou. Proto je vhodné považovat CAH za kontinuum fenotypů, od těžkých po mírné nebo jinak asymptomatické (3).
nejběžnější formou CAH je způsobeno tím, že 21-hydroxylázy deficit (21OHD), což má za následek zhoršenou nebo ne konverze z 17 hydroxyprogesterone (17 OHP) na 11 deoxycortisol a progesteronu deoxykortikosteronu. Blokáda steroidní výsledky konverze při zvýšené produkci androgenů prekurzorů, pod CRH-ACTH stimulaci, což vede k biochemické hyperandrogenism, která se vyznačuje zvýšenou 17-OHP úrovně. Klasická forma onemocnění se vyskytuje u 1 ze 16 000 živě narozených dětí po celém světě. Je však třeba zmínit, že vnímaná incidence onemocnění souvisí s použitou screeningovou metodou. Například, bylo zjištěno, že výskyt klasické formy CAH ve Švédsku je 1:11,500, když se případ průzkum přístup je používán, který údaj je však klesne na 1:9,800, když hormonální screening je aplikován. Podobně, výskyt ve Francii a Švýcarsku se pohybuje mezi 1:15,472 a 1:23,000 a 1:10,749 a 1:11,661 při různých metod screeningu jsou používány (4).
NCCAH je mnohem častější, vyskytující se přibližně u 1 z 1 000 Bělochů a častěji u určitých etnických skupin, jako jsou Ashkenazi Židé (1:27), Hispánci (1:53), Jugoslávci (1:62), a Italové (1:300) (5, 6). Nicméně údaje o prevalenci NCCAH založené na různých metodách odhadu (případový průzkum, hormonální screening a molekulární testování) chybí. NCCAH je považována za nejčastější autozomálně recesivní endokrinní poruchu s nosnou frekvencí 1: 25 až 1: 10. V NCCAH v důsledku 21-OHD se zbytková enzymatická aktivita odhaduje na přibližně 10-70% (7-9).
Diagnostika
Ve srovnání s diagnózou klasická forma onemocnění, která je vyrobena při narození nebo v novorozeneckém období, protože genitální nejednoznačnost a/nebo soli-plýtvání příznaky nebo prostřednictvím screeningové programy jsou využívány v některých zemích většině případů NCCAH nejsou snadno zjistitelné (4, 10). Navíc, mnozí jedinci zůstávají bez příznaků v dětství a dospívání, mají normální reprodukční funkci, a pouze se o NCCAH vzhledem k diagnóze jiného člena rodiny a následné testování (11). Nicméně, většina žen s NCCAH vyhledat lékařskou pomoc, když se objeví příznaky androgen nadbytku a při klinickém podezření vyzve testování, zvýšené bazální 17 OHP úrovní bude více pravděpodobné než ne přejděte na diagnostiku NCCAH.
klinické pokyny navržené endokrinní společností skutečně doporučují základní nestimulovanou hodnotu 17 OHP jako screeningový nástroj pro NCCAH. Ráno 17 OHP v séru >6 nmol/L ve folikulární fázi u menstruujících žen by měla být podnětem k dalšímu hodnocení, neboť bylo prokázáno, že hladiny nad 6 nmol/L zachytit 90% NCCAH jedinců (12, 13). Náhodná měření 17 OHP se neprokázala jako užitečná, protože tato často poskytují normální hladiny u pacientů s NCCAH a navíc jsou extrémně vysoké v luteální fázi u poloviny zdravých žen (13). Nicméně, v našich datech odvozených od 280 subjektů s onemocněním, šest pacientů (2 .1%) měla výchozí hodnotu 17 OHP < 6 nmol/L (14). Dodatečně, Bidet et al. ve velké kohortě žen s NCCAH ověřena molekulárními technikami, také zjistil, bazální 17 OHP hodnoty nižší než 6 nmol/L u 8% pacientů studovány (15). Konečně, na základě údajů shromážděných Speiser et al., 9% jedinců s NCCAH zobrazilo 17 hodnot OHP nižších než 2 ng / ml (což odpovídá 6nmol/L). Podle jiných studií postačuje výchozí hodnota 17 OHP mezi 5, 1 a 9 nmol / L pro diagnózu NCCAH (13 ,16, 17). V poslední době, úroveň bazální 17 OHP 4.6 nmol / L byl navržen jako práh pro ACTH testování k predikci NCCAH u subjektů s předčasným adrenarche během dětství (18).
součet těchto nálezů a návrhů naznačuje, že výběr pacientů, kteří podstoupí stimulační test Synacthen, by měl být hodnocen případ od případu. Pro diagnózu je vyžadována hladina 17 OHP po stimulaci nad 3 nmol/L (19). Heterozygoti nesoucí jednu mutaci CYP21A2 vykazují po stimulaci ACTH mírně zvýšené hladiny 17 OHP, i když u neovlivněných subjektů dochází k překrývání (9). Nicméně, jak Dacou-Voutetakis et al. poukázali na to, je-li součet bazálního a post-stimulace 17 OHP hodnoty větší než 1,5 nmol/L, pak možnost heterozygotnost je mimořádně vysoká (20).
další důležitá úvaha se týká technik používaných pro hodnocení 17 OHP. 17 hladin OHP se měří různými metodami imunoanalýzy, ale jak se nedávno ukázalo, nejpřesnější a nejspolehlivější výsledky byly dosaženy implementací kombinace kapalinové chromatografie s hmotnostní spektrometrií (LC-MS/MS). Opravdu, mnoho falešně pozitivní 17 OHP měření byly nalezeny při LC-MS/MS měření byly porovnány se standardními metodami (21) Nicméně, druhý postupy nejsou všeobecně používané, v případě nejisté diagnózy, budou poskytovat přesnější výsledky. Je třeba poznamenat, že screeningové a diagnostické prahové hodnoty pro LC-MS/MS kvantifikované 17 OHP ještě nejsou definovány. Tyto prahové hodnoty budou pravděpodobně nižší než prahové hodnoty stanovené imunoanalýzami kvůli zvýšené specificitě LC-MS/MS, což je metoda méně náchylná ke zkřížené reaktivitě a interferencím (22, 23). Zda je pro definitivní diagnózu vyžadován profil steroidů v moči, je třeba objasnit (22). V hraničních případech, je vhodné získat kompletní adrenokortikální profil po ACTH stimulační test k odlišení 21-hydroxylázy deficit z jiných enzymu vady a stanovit pevné diagnózy.
konkrétně by měl být v nejednoznačných případech proveden kompletní steroidní profil, aby se potvrdil deficit 21-hydroxylázy a vyloučily se další enzymatické detekce. Zařazení 17 OHP, kortizol, 11-deoxykortikosteronu,11-deoxycortisol,17-OH-pregnenolon, Dehydroepiandrosteron a androstendion v séru steroidní profil by být užitečné k vyloučení jiných příčin CAH jako 11beta-hydroxylázy deficit a, více zřídka, 3β-hydroxysteroidů dehydrogenázy nebo P450 oxidoreduktázu nedostatek (24). Zatímco genetické testování se v tuto chvíli nepovažuje za primární diagnostický nástroj pro NCCAH, je povinné pro potvrzení diagnózy a pro genetické poradenství (3).
pokud jde o hladiny kortizolu, obecně se očekává post-stimulační hodnota alespoň 496 nmol/L. Za zmínku stojí, podle Stoupa et al. 60% 47 dětí s NCCAH výsledkem 21 OHD měl nízké hodnoty kortizolu po stimulaci, zjištění ukazuje na potřebu zvýšené ostrahy pro rozvoj nedostatečnost nadledvin během hlavní stresor události (25).
NCCAH Projevy u Žen
klinické výraz NCCAH se vyznačuje vysokou úrovní polymorfismu, co se týká nejen věk nástupu, ale také různé příznaky a symptomy. Uvádí se, že první klinická prezentace NCCAH je v 11% případů před dosažením věku 10 let a v 80% ve věku 10 až 40 let (12). Korelace genotyp-fenotyp v CAH a NCCAH dosud nebyla objasněna. Speiser a kol. naznačují, že většina, ale ne všechny fenotypové variability deficitu 21-hydroxylázy je výsledkem alelické variace CYP21A2 (26).
projevy v dětství
v novorozeneckém období zůstávají samice NCCAH obvykle asymptomatické a mají normální vnější genitálie. Nejčasnější případ nccah hlášeny je 6 měsíců stará dívka, která vyvinula ochlupení (11). Klinické nálezy a příznaky v případech NCCAH obvykle začínají ve věku 5 let nebo dokonce později a souvisejí se zvýšenými hladinami androgenu, i když v některých případech může dojít k mírnému nedostatku kortizolu (8). V 92% NCCAH případech, prvním příznakem, který se projevuje, je předčasnou pubarche (PP), který se vyskytuje pod 8 let věku u dívek a 9 lety u chlapců (12) s odhadem prevalence mezi 10. a 11.3% (15). Je třeba poznamenat, že u dětí uvedených s PP se podle různých studií incidence NCCAH pohybuje od 5 do 30% (20, 27). Na základě naší analýzy 280 pacientů s molekulárním potvrzením NCCAH jsme však zjistili, že incidence PP u 94 žen mladších 8 let byla až 88%. Na druhé straně analýza 45 mužů s NCCAH identifikovala PP pouze u 29% subjektů (14). Dále bychom měli mít na paměti, že asi polovina subjektů s PP může být heterozygotními nosiči mutace CYP21A2.
dalším aspektem, který vyžaduje objasnění, je vztah mezi PP a předčasnou pubertou. Předpokládá se, že v některých případech může periferní aromatizace adrenálních androgenů na estrogeny aktivovat osu hypotalamus-hypofýza-gonadální, což vede k sekundární předčasné pubertě (28). V takovém případě je zapotřebí pravidelné sledování. U některých dětí se rozvíjí hirsutismus, akné a / nebo rychlý růst a projevuje se vysoká struktura a vývoj kostního věku (29). Ten může mít za následek zkrácenou konečnou výšku v důsledku rychlé epifyzální fúze. Mezi další vzácné klinické nálezy během dětství patří labiální adheze, perianální vlasy, clitoromegalie a chrapot hlasu (30-32).
Projevy v Dospívání a dospělosti
Během dospívání a dospělosti, NCCAH u žen představuje s hyperandrogenních příznaky připomínající syndrom polycystických ovarií (PCOS) (33). Mezi příznaky patří hirsutismus, akné, androgenní alopecie, anovulace, menstruační dysfunkce a neplodnost. V multicentrické studii byly nejčastějšími příznaky u dospívajících a dospělých žen hirsutismus (59%), oligomenorea (54%) a akné (33%). Mezi 161 ženy s NCCAH, prezentující příznaky jsou hirsutismus (78%), menstruační dysfunkce (54.7%) a snížená plodnost (12%) (34). V naší skupině 161 žen s onemocněním 63% pacienta prezentovány s polycystických vaječníků podobný fenotyp (14). Toto zjištění má opačný dopad, protože incidence NCCAH je vysoká u žen s hyperandrogenemií. Jistě, studie Witcel et al. hlášení incidence NCCAH v rozmezí od 1 do 33% u hyperandrogenních žen je velmi zajímavé. V jiné studii 950 žen, které byly předány k hodnocení nadbytku androgenů, byl nccah zjištěn u 4,2% z nich. Pacienti s NCCAH však byli mladší a chlupatější ve srovnání s ostatními skupinami hyperandrogenních pacientů. Byly také charakterizovány významně vyššími hladinami testosteronu, volného testosteronu a 17 OHP než pacienti s jinými hyperandrogenními syndromy. Při ovariálním ultrazvuku vykazovalo 77% z nich polycystické vaječníky a 41% zvýšilo velikost vaječníků. Celkově splnily kritéria PCOS podle NIH a Rotterdam v procentech 56 a 72,8% (35).
progresivní povaha onemocnění je zdůrazněna skutečností, že prevalence hirsutismu se s věkem zvyšuje a bylo pozorováno, že je vzácná před pubertou. Hirsutismus je nejčastějším klinickým projevem hlášena u pacientů s NCCAH, v rozmezí od 71 do 96% (28, 36). Pubertální dívky s NCCAH obvykle vykazují hirsutismus (37, 38). Na druhém konci spektra je otázka alopecie. U pacientů s NCCAH je hlášeno plešatění mužského vzoru. Prevalence alopecie se také zvyšovala s věkem, ze 6% u pacientů během druhé dekády jejich života na 19% v páté, což opět naznačuje progresivní povahu onemocnění (39). Akné je hlášeno u téměř 33% subjektů NCCAH (36). Je pozoruhodné, že mutace genu CYP21A2 způsobujícího NCCAH byly detekovány u 4, 9% z 123 dospělých žen s těžkým akné (40).
Menstruační nesrovnalostí včetně oligomenorhey nebo sekundární amenorea může být často představují znamení NCCAH v post menarche jedinců. Navíc 8-9% případů také zažívá primární amenoreu, a proto poprvé vyhledejte lékařskou pomoc. Navíc u jedinců NCCAH je oligomenorea častější než primární amenorea. Například v sérii od Moran et al. mezi NCCAH žen ve věku 10 až 19 let, oligomenorhey byl přítomen v 56% případů, ve srovnání s pouze 9% z dospívání s primární amenorea (12). Na závěr, tolik NCCAH příznaky se podobají těm PCOS, že to není překvapující, že NCCAH byl nazván velký mimicker PCOS. V každém případě by měla být při hodnocení jakékoli mladé ženy, která je označována za hyperandrogenní příznaky, zvážena diagnóza NCCAH.
ve studii Arlt et al., pacienti s NCCAH byli charakterizováni významně vyšším BMI a nižší konečnou výškou ve srovnání s odpovídajícími kontrolami (41). Tyto údaje, společně s několika studií, které ukazují na přítomnost inzulínu necitlivost, naznačují nepříznivý metabolický profil, který by mohl být podrozdělen vysvětlil tím, že účinek zvýšené koncentrace androgenů a/nebo v důsledku terapie glukokortikoidy (42, 43). Osteoporóza může být také detekována u těchto subjektů, pravděpodobně v důsledku léčby kortikosteroidy (34).
NCCAH Vedení Z První Projev na Dospělý Život
Jakmile je diagnóza NCCAH bylo zjištěno, několik otázek týkajících se následujících doporučení léčby příkaz úvahu. První otázkou, kterou je třeba řešit, je, zda je indikována léčba glukokortikoidy (GC). Obecné důvody tohoto přístupu je, že tím, že poskytuje dostatečné koncentrace kortizolu pro pacienta, její denní potřeby jsou pokryty, a, v důsledku toho, CRH-ACTH osy stimulace bude zúžený, což vede ke snížení adrenální produkci androgenů. Obecné pravidlo týkající se tohoto přístupu je, že Gc jsou uvedeny na výměnu a ne farmakologických dávkách, zatímco vliv věku, pohlaví, laboratorní údaje pacienta-konkrétní doporučení a cíle léčby na být náhradní léčba glukokortikoidy jsou vážně brány v úvahu. Nicméně, měli bychom také být vědomi toho, že dlouhodobé GC substituční terapie může vést k hypercortisolemia, s výslednou dobře zdokumentované nepříznivé účinky na každý aspekt metabolismus, a to zejména růst, distribuci tuku a inzulínové rezistence, stejně jako na psychologický profil. Další hlavní nevýhodou tohoto přístupu je nedostatek adekvátního klinického indexu nebo biochemického markeru adekvátní náhradní dávky, jako jsou například hodnoty TSH u hypotyreózy.
a Konečně, skutečnost, že neexistuje shoda, jak na GC, které by měly být použity, a absence dlouhodobé údaje týkající se různých způsobů GC správy dále komplikovat ošetřujících lékařů rozhodování a výběr optimální výběr pro každého pacienta. Hydrokortizon je typicky používán u dětí, stejně jako to se nejvíce podobá přirozenému hormonu (kortizolu), ale to není považováno za vhodný přístup u dospívajících a mladých žen z důvodu potřeby více denní dávkování. Proto většina dospělých endokrinologů preferuje buď dexamethason nebo prednisolon ve vhodných dávkách. Na druhé straně je třeba zdůraznit, že rovnocennost různých GCs je založena na jejich protizánětlivém účinku a nikoli na různých aspektech lidského metabolismu. Protože kortizol postihuje téměř 20% lidského genomu, očekávají se různé reakce různých GCs v různých tkáních. Potvrzením tohoto vnímání je skutečnost, že podávání dexamethasonu pacientům s CAH prokázalo zhoršení indexů inzulínové rezistence ve srovnání s jinými GCs (44). Navíc podávání 2,5-7,5 mg prednisolonu, dávky považované za normální, má dlouhodobý negativní dopad na metabolismus kostí (45). Proto by měl být hydrokortizon považován za nejlepší formu léčby v případech suplementace GC. Příchodem nově syntetizované hydrokortison vzorec s jedním pilulku za den a jeho počáteční pozitivní výsledky u pacientů s CAH ukazuje velký příslib pro budoucnost (46).
Řízení V Dětství
NCCAH pacientů, kteří jsou diagnostikovány během dětství s příznaky PP mohou být léčeni kortikoidy s cílem potlačení nadledvin hormony a brání rychlému rozvoji kostní věk, který by mohl mít vliv na konečné výšky. U pacientů, u kterých byla léčba hydrokortizonem zahájena 1 rok před nástupem puberty a kteří měli kostní věk nižší než 9 let, zůstala konečná výška v rámci genetického potenciálu (47). Dostupné studie naznačují, že výška v dospělosti přiblížil očekávané cílové výšky u pacientů, které byly pečlivě sledovány a kteří striktně dodržovány léky plány (48-50). Nedávné studie malé pěti případech (u pacientů 6.1–9.2 let) prokázala, že ultranízkých dávky dexamethasonu je slibnou možností, jak snížit endogenní stres a jeho účinky. Zda to představuje realistický přístup a kolik by měla být podaná dávka, je třeba objasnit (51). Studie Nebesio et al. porovnával hormonální účinky a farmakokinetiku hydrokortizonu, prednisolonu a dexamethasonu u 9 prepubertálních pacientů s CCAH. Ty ukázaly, že dexamethason byl silnější, že jiné formy v dosažení výrazně nižší hladiny hormonů, proto naznačuje, že dexamethason je nejúčinnější pro supresi adrenální produkci androgenů (46). K ověření nebo odmítnutí tohoto zjištění jsou samozřejmě zapotřebí další studie. Navíc v některých případech mohou zvýšené koncentrace androgenu vést k sekundární stimulaci osy GnRH, což vede k předčasné pubertě. V takovém případě paralelní kurz s GnRH analog může být předepsán, pokud kostní věk je výrazně vyšší než chronologického věku a/nebo předpokládané konečné výšky je nepřiměřené cílové výšky.
Další důležité úvahy týkající se případů souběžného nedostatku GH v případě, že léčba analogy GnRH a předpisem GH vedla k dosažení cílové výšky (52). Podle stávajících pokynů, výše popsané terapie je opatrně doporučuje pouze pro ty případy, v nichž předpokládaná výška SD je -2.25 < cílové výšky, nebo dokonce nižší (53). Alternativní indikací pro zahájení léčby hydrokortizonem je nedostatečná odpověď kortizolu po stimulaci ACTH (36).
preferovanou léčbou GC u dětí je obvykle hydrokortizon 10-15 mg / m2, rozdělený do tří dávek. Často se ukázaly jako účinné i nižší dávky, počínaje 6 mg / m2 / den (34). Předávkování je třeba se vyhnout, s ohledem na to, že může mít za následek špatný růst, stejně jako Cushingoid funkce. Pravidelný růstový vzorec, kostní věk kompatibilní s chronologickým věkem, a absence centrální obezity může také sloužit jako klinické indexy pro vhodné řízení. Pokud jde biochemické/ hormonální profil, androstendionu a testosteronu v polovině až horní rozsahy pro kostní věk jsou považovány za lepší značky odpovídající GC substituční terapie u dětí s NCCAH. Nicméně, potlačení hladiny testosteronu byly nalezeny v 10% NCCAH pacientů, vzhledem k tomu, dalších 28% pacientů mělo zvýšené koncentrace testosteronu, tento jev může být připadající na hydrokortizon variabilita (54, 55). Hodnoty DHEAS se snižují s velmi malými dávkami, zatímco potlačení 17 hladin OHP a progesteronu vyžaduje velmi vysoké dávky GC. Je třeba poznamenat, že 17 hodnot OHP je pro diagnózu rozhodující, ale během sledování není užitečné. Je třeba poznamenat, že odběr krve pro hormonální hodnocení musí být proveden bez ukončení léčby. Vzhledem k nejasnosti onemocnění a jeho mnohostranné povaze však neexistují žádné specifické pokyny pro načasování změn režimu nebo ukončení léčby glukokortikoidy u dětí.
Řízení Během Dospívání
Pacientů Léčených Od Dětství
Až do vzniku normálního menstruačního vzor v NCCAH holky, pokračování GCs, která začala v dětství, je vysoce doporučeno (56). Dospívající pacienti však často nevykazují dostatečnou shodu s chronickým podáváním léků a často vynechávají dávky. Navíc, během puberty, half-life hydrokortison klesne o 50% v důsledku zvýšené hladiny IGF-1, což snižuje 11ßOHSD činnosti, jakož i v důsledku zvýšeného kortizolu vůle pramenící z amplifikace glomerulární filtrace (57). U mladé ženy, 2 roky po menarche a pokud byly zaznamenány normální ovulační cykly, důrazně se doporučuje přístup zaměřený na pacienta k hyperandrogenním příznakům, které se mohou objevit.
Pokud existují závažné hyperandrogenních zjištění, jako je hirsutismus, akné a/nebo oligomenorhey, pokračování léčby je třeba zvážit. Je důležité si uvědomit křehkou citlivost mladého adolescenta, který bude vážně poškozen „odpuzujícími“ příznaky hyperandrogenismu. Kombinace hyperandrogenních příznaků, menstruačních poruch a špatné kvality života je dobře zdokumentována a je zvláště vysoká u mladších žen. Dalším bodem, který je třeba zvážit, je reakce adrenální rezervy po stimulaci ACTH. Vzhledem k tomu, že vrchol kortizolu u pubertálních a dospělých žen po stimulaci je nižší než 496 nmol / L, v případě stresu by měla být podána léčba steroidy (58). Na druhou stranu, pokud se u mladého pacienta neobjeví žádný z výše uvedených příznaků, může být léčba GC přerušena, po které se doporučuje pravidelné sledování.
Samice S Prvním Projevem Během Dospívání nebo Hyperandrogenních Příznaky Po Ukončení Léčby,
V přítomnosti hyperandrogenních příznaky, pacient orientovaný přístup je vysoce doporučeno, se zaměřením na hlavních stížností pacienta. To je mnohem lepší než obecný univerzální modus operandi, protože s druhým přístupem bude mladý pacient zklamán, což povede k přerušení léčby a v důsledku toho k nepříznivým zdravotním výsledkům v dospělosti. V případech s těžkým hirsutismus, během 6 až 12 měsíců s perorální antikoncepce (Antikoncepcí) představuje přiměřený postup vzhledem k blahodárné účinky Antikoncepcí na SHBG v játrech výroby a poklesu androgenů uvolnění z vaječníku. Klinické zlepšení bude očekáváno po nejméně 2-3 měsících zahájení OCPs, zatímco současné užívání progestagenu s minimálními androgenními vlastnostmi je vysoce doporučeno. Pokud OCP selžou jako první linie, mohou být k léčbě přidány antiandrogeny (spironolakton, flutamid, bikalutamid, cyproteronacetát a finasterid). Podle našich zkušeností, podávání bikalutamidu bylo dosaženo významného zlepšení v případě těžké akné, ale podobné výsledky byly získány v případech s těžkou hirsutismus. Kosmetické přístupy, jako jsou laserové aplikace a depilační přípravky mohou být také navrženy pro ženy si stěžují na nadměrné nebo nežádoucí růst vlasů nebo pokožky hlavy vypadávání vlasů (androgenní alopecie) (32, 34, 57).
u pacientů s nedostatečnou sekrecí kortizolu po stimulaci nebo v případě, že OCP a antiandrogeny nemohou být tolerovány, je vysoce doporučen průběh s GCs (57). Data z New et al. uveďte, že nepravidelná menstruace a akné jsou obráceny do 3 měsíců po zahájení léčby glukokortikoidy, zatímco hirsutismus vyžaduje téměř 30 měsíců (59). Na rozdíl od dětství se v dospívání často používají déle působící steroidy a doporučují se režimy 5 mg prednisolonu nebo 0,25 mg dexamethasonu (53). V reálném světě však klinické údaje ukázaly řadu různých režimů používaných při léčbě NCCAH (41).
primárním cílem léčby by měla být pacientova úleva od hyperandrogenních symptomů. Tedy u jedinců s NCCAH diagnostikována pouze na základě laboratorních výsledků, klinické funkce by měla vést řízení doporučení, protože hormonální a molekulární nálezy nemusí nutně předvídat klinické závažnosti. Podle pokynů Endokrinní společnosti by pacienti s NCCAH měli mít možnost přerušit léčbu GC, jakmile příznaky vymizí (60). Tito pacienti by samozřejmě neměli být ztraceni při sledování, zatímco léčba by měla být znovu zahájena v případě opětovného výskytu příznaků. Dále v případě přerušení léčby by pacienti měli být informováni o možnosti neplodnosti a měli by být povzbuzováni, aby vyhledali lékařskou pomoc, pokud si přejí otěhotnět (19). Je třeba poznamenat, že vhodný přenos pacienta z pediatrického na dospělého endokrinologa by měl být proveden optimálně po 1 roce synchronizovaného sledování (53, 61).
NCCAH a Reprodukce
Subfertility
Subfertility je často hlášena u klasické formy CAH, což je způsobeno především menstruační poruchy, chronické anovulace, a anatomické deformity. Míra porodnosti byla odhadnuta na 17% ve srovnání s 65% kontrolní populace (62). Mezitím byly údaje týkající se plodnosti u žen s NCCAH nedávno podrobně posouzeny a odhadovaná incidence neplodnosti je 11% u žen NCCAH, tj. Bidet et al. hodnotila plodnost u 190 žen trpících NCCAH, z nichž 95 chtělo otěhotnět. V této populaci bylo hlášeno 187 těhotenství u 85 žen, což mělo za následek 141 porodů u 82 jedinců. Je třeba zdůraznit, že 99 těhotenství (52.9%) došlo před diagnózou NCCAH, tři z nich s aplikací indukce ovulace protokoly, zbytek je spontánní, zatímco 88 těhotenství došlo po NCCAH diagnózu. Ve velké většině z nich se těhotenství vyvinulo po zahájení léčby hydrokortizonem, zatímco u 11 žen se to stalo spontánně (63, 64). V jiné zprávě o 22 pozorovaných ženách NCCAH, které si přejí těhotenství, následovalo 12 těhotenství s prednisonem (65).
potraty
v kohortě bidetu et al., míra potratu byla 6,5 a 26,3% u pacientů léčených GCs a neléčených pacientů (64). Výsledek těhotenství může ještě být úspěšný bez jakékoliv glukokortikoidní léčby v případech, kdy NCCAH ještě nebyla diagnostikována, jak je uvedeno také v případě, že zpráva Cuhaci et al. Stejný pár však zažil dva potraty a po tomto prvním těhotenství hlásil subfertilitu (66). Pokud jde o možnost předčasného těhotenství, jak hodnotí Bidet et al., se výrazně neliší od zbytku populace (15).
plánování plodnosti
u žen se symptomatickým hyperandrogenismem nebo s hlášenou neplodností, ale které si přejí otěhotnět, se důrazně doporučuje GC terapie. Během tohoto období jsou užívanými glukokortikoidy prednison nebo hydrokortizon. Nejdůležitější akcí, protože bude řídit následující kroky, je genetické testování potenciálních rodičů. Ženy s NCCAH, které si přejí otěhotnět, by si měly být vědomy rizika porodu dítěte s klasickou formou onemocnění. Výsledek těhotenství u žen s NCCAH a konkrétněji výskyt kojenců narozených s CCAH se v nedávných studiích odhaduje na 1, 5 až 2, 5%, s NCCAH přibližně 15% (36, 64). Tato frekvence samozřejmě závisí na referenční populaci, což je vyšší výskyt u populací s vysokou mírou sňatku (36). Předpokládané šance na rodiče porodila dítě s CAH je 1: 120 pro neznámé otcovský genotyp, nula v případě, že otec nemá žádné relevantní mutace, a 1: 4, když má heterozygotní mutací (19). Výsledkem je, že genetické testování partnerů těchto žen je nezbytné pro posouzení rizika porodu dítěte s klasickou formou CAH (64, 67).
V případě, že budoucí matka nese dvě NC mutace (Tabulka 1), není tam žádný absolutní nutnost pro genetické testování se v budoucnu otcem, protože, pokud je heterozygotní pro CYP12A2 mutace, potomci, je riziko vzniku non-klasické formy onemocnění v budoucnosti. Je však třeba poznamenat, že stav nosiče je 2-15% v různých populacích a polovina těchto jedinců nese závažnou mutaci. Dále, definitivní doporučení týkající se situací, kdy není vyžadováno genetické testování, jsou obtížná vzhledem k nedokonalé korelaci genotyp-fenotyp, zejména pro mírnější mutace . Například mutace P30L, nejčastěji spojená s NCCAH, se vyskytuje také u pacientů s klasickým CAH . Velká multicentrická evropská studie nedávno ukázala, že tomu tak bylo u 78% pacientů . V této studii byla mírná mutace V218L spojena spíše s klasickou než NCCAH ve 30% případů. Proto by všem pacientům s NCCAH mělo být nabídnuto genetické poradenství a molekulární hodnocení reprodukčních partnerů.
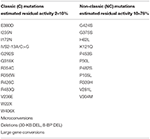
Tabulka 1. Mutace CYP21A2 s minimální (C) nebo střední (NC) reziduální enzymovou aktivitou (14, 68, 69).
naopak, v případě, že budoucí matka je složený heterozygot s jednou těžkou (C) a jeden NC mutace, pak genetické testování budoucího otce je povinné. Měli bychom mít na paměti, že data pocházejí ze studií s použitím imunoanalýzy nebo přesnější LC-MS/MS postupy, důsledně upozornit na neschopnost spolehlivě vyloučit heterozygotnosti pomocí bazální nebo ACTH stimuluje 17 OHP hodnoty, vzhledem k výraznému překrývání s normálním rozmezí. Jeden může navrhnout použití Synacthen test, a pokud to není kompatibilní s heterozygotnost (součet hodnot bazální a maximální stimulované 17 OHP hodnoty < 1.5 nmol/L), pak testy DNA by být vyhnout. Při interpretaci tohoto testu bychom však měli být velmi opatrní, protože v jiných studiích nebyl validován. Proto, použití ACTH stimuluje 21-deoxycortisol buď samostatně, nebo jako součást steroid poměr nebo steroidní profil, může usnadnit biochemické identifikace heterozygoti v budoucnu, zejména v LC-MS/MS se stává více široce dostupné . Pokud budoucí otec nese mutaci NC, pak není potřeba nic jiného. Naopak, pokud je nositelem klasické mutace, vyvstane otázka týkající se prenatální léčby plodu. Všechny možné kombinace genotypů a navrhované postupy jsou uvedeny v tabulce 2.

Tabulka 2. Plánování navrhovaných postupů během těhotenství na základě genotypu potenciálních rodičů.
Prenatální Léčba CAH
cílem prenatální léčby CAH je prevence pohlavních virilizace plodu, ale také zmírnění stresu, pociťují rodiče, kteří jsou pravděpodobné, že mít dítě s obojetným genitálem (70). Dexamethason se používá, protože jeho schopnost procházet placentou, a proto to není deaktivován z placentární 11beta – steroid dehydrogenázy a váže jen minimálně do krve matky kortizol vázající globulin (CBG). Dexamethason potlačením sekrece ACTH plodu snižuje zvýšenou produkci androgenu plodu. Léčba musí být přerušena v případě, že analýza karyotypu nebo DNA odhalí muže nebo neovlivněnou ženu (71). Nicméně, i když plodu genitální virilizace již začala na 6-7 týdnů po početí, choriových klků biopsie pro genetická diagnostika nemůže být získán před 10.−12. týden. Tento časový interval naznačuje, že všechny rizikového těhotenství pro virilizující CAH by měly být léčeny, i když pouze 1 z 8 plodů je ovlivněna a žena (19, 34, 70).
zda je tato expozice plodu dexamethasonu pro preventivní opatření lékařsky a eticky přijatelná, zůstává kontroverzní (70). Studie na zvířatech prokázaly, že v prvním trimestru dexamethason snižuje porodní hmotnosti, ovlivňuje ledvin, slinivky funkce beta-buněk a vývoj mozku, a předurčuje, úzkost, hypertenze a hyperglykémie v dospělosti (70). Kromě toho 10-20% těhotných žen užívajících léčbu dexamethasonem během těhotenství si stěžuje na přírůstek hmotnosti, zvýšenou chuť k jídlu, změny nálady, nespavost, otoky, hypertenzi, hyperglykémii a strie. Nicméně, nový et al. našel nižší než průměrná Prader skóre u plodů léčených s dexamethasonem prenatálně, ale žádný rozdíl v dlouhodobých výsledcích (72). Dodatečně, v recenzi Merce Fernandez-Balsells et al. ukázalo se, že Dexamethason je spojen se snížením virilizace plodu bez významných nežádoucích účinků na matku nebo plod (63). Nicméně, autoři výše uvedené recenzi, stejně jako po sobě jdoucí pokyny Endokrinní Společnosti poukazují na to, že vzhledem k malé velikosti vzorku v celém těle literatury, téma zůstává nejistá a další vyšetřování je nezbytně nutné.
rozhodnutí o zahájení léčby by měly být prováděny pouze ve velkých centrech se zkušeným týmem a protokoly schváleny Institucionální Recenze Desek a na základě rodinné hodnoty a preference a s jejich písemný informovaný souhlas jako předpoklad (19, 63). Několik specialistů v oblasti naznačují, že vyšší hodnoty by měl být kladen na prevenci zbytečných prenatální expozice matky a plodu na dexamethason, spíše než o uložení emocionální mýtného nejednoznačné genitálie na rodiče a pacienty. A co je nejdůležitější, mělo by být rodičům objasněno, že podávání dexamethasonu nemění stav pacienta, ale je zaměřeno spíše na snížení potřeby chirurgického zákroku než na zachování života nebo intelektuální kapacity. Musí také vědět, že pravděpodobnost, že jejich dítě bude trpět klasickou formou onemocnění, je navzdory léčbě vysoká. Nejvíce rozhodující, mezitím, expozice mladý organismus velmi silný GC během obzvláště citlivé období fetální programování a růstu, což by mohlo ukázat jako zbytečné, v případě, že plod s XY karyotyp, není v současné době podporován robustní a nezpochybnitelná data.
V ideálním případě by měla existovat včasná diagnóza, která se provádí neinvazivní technikou a před začátkem genitální virilizace plodu. Pracovat v tomto směru, nové studie ukazují na použití cell-free fetal DNA získané z mateřské plazmy jako slibná metoda, která umožní stanovení fetálního pohlaví a diagnóza CAH v rané gestační věk (<9 týdnů). Pokud je tento postup široce implementován v klinické praxi, zabrání se zbytečné prenatální léčbě dexamethasonem (73).
Léčba Během Těhotenství
Během těhotenství, ženy s NCCAH by měl být přednostně léčeni kortikoidy, a to nejen kvůli těhotenství, je obvykle doprovázeno zvýšenou hladinu stresu, ale také jako preventivní opatření proti výše uvedeným vysokým výskytem potratů. Tato forma glukokortikoidu je upřednostňována díky jeho metabolizaci placentárním enzymem 11β OH steroidní dehydrogenázou II, která zabraňuje glukokortikoidu mít jakýkoli účinek na plod (71). V době početí by měla být dávka hydrokortizonu zvýšena na 20-25 mg / den a modifikace dávky se provádějí každých 6-8 týdnů s cílem udržet hladiny testosteronu na horních normálních hladinách trimestru těhotenství (74). Jako příklad jsou uvedeny hodnoty testosteronu a D4 pacienta s NCCAH od početí do 3 let po porodu dvou zdravých dvojčat(Obrázek 1).
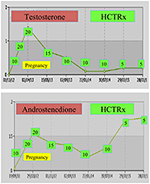
Obrázek 1. Hladiny testosteronu a andostendionu a následné dávkování hydrokortizonu (HCTRx) od početí do 3 let po porodu podávané pacientovi s NCCAH.
Další Speciální Otázky
zvládání Stresu v NCCAH
Během hlavní život ohrožující stres, operace, nebo vážné nemoci, pacienti s NCCAH, kteří jsou glukokortikoidy-léčen, může vyžadovat větší nebo častější dávky glukokortikoidů vzhledem k tomu, že jejich funkce nadledvin je iatrogenically potlačena. Je proto zásadní vzdělávat rodiče malých dětí a také převychovávat pacienty o jejich přechodu na péči o dospělé, o dávkování stresu. U pacientů s NCCAH, kteří nejsou léčeni GCs nebo v případě přerušení léčby, by měla být hodnocena kortizolová odpověď na ACTH. Téměř třetina pacientů s NCCAH reaguje na stimulaci nedostatečně (15, 75). U těch, kteří normálně reagují na stimulaci ACTH, se nedoporučuje léčba dávkami stresu (19). Léčba mineralokortikoidy se v žádném případě nevyžaduje, protože tito pacienti mají normální sekreci aldosteronu a nevyvíjejí plýtvání solí (58). Poznámky, v případě hypertyreózy nebo zvýšení levothyroxin léčbu, clearance kortizolu je zvýšená a adrenální krize může nastat (58, 76).
Psychobiologické aspekty v NCCAH
Meyer-Bahlburg et al. v několika studiích bylo hlášeno zhoršený psychologický profil u pacientů s NCCAH kvůli nedostatku 21OH. Přesněji řečeno, postižené ženy měly vyšší maskulinizace/defeminization skóre na několika opatření z genderového chování ve srovnání s běžnou kontrolu ženy, i když výrazně méně než u žen s klasickým CAH. Rovněž měli výrazně zvýšenou bisexuální nebo homosexuální orientaci. Retrospektivní klinicko-kvalitativní rozhovory s těmito ženami odhalila historii nepohodlí a sociální stres související s jejich pre-léčba, zkušenosti s androgen-dependentní příznaky, jako je akné, hirsutismus, a pojetí obtíží (59, 77, 78). Podobně, Arlt et al. ukázal, že subjektivní zdravotní stav byla výrazně narušena ve všech osmi doménách krátkodobého průzkumu zdraví, s nejvýznamnější rozdíly ve srovnání s věkem a pohlavím odpovídající kontroly, které se týkají domén, všeobecné zdraví, vitalita, a roli omezení vzhledem k citové problémy. Dotazník nemocniční úzkost a deprese (HADS) také odhalil zvýšené skóre úzkosti (41). Berouce v úvahu tato zjištění, psychologické parametry pro vedení terapie by měla být považována u žen s NCCAH a v rámci přístupu orientovaného na pacienta, psychologické diagnostiky a podpora musí být nabízeny.
závěry
NCCAH je považován za častější a mírnější formu CAH kvůli retenci 20-50% enzymové aktivity. Většina pacientů může vyhledat lékařskou pomoc v jakékoli fázi svého života kvůli příznakům souvisejícím s nadbytkem androgenu. Patří mezi ně předčasné pubarche, rychlý růst, hirsutismus, akné, menstruační nepravidelnosti nebo neplodnost, stejně jako mnoho dalších méně významných projevů. Brzy ráno výchozí hodnoty 17 OHP jako dobrý počáteční screeningový test a další hodnocení se stimulací ACTH a v případě hraničních výsledků se doporučuje genetické testování. Obecně platí, že během sledování by se měl používat androstendion a ne 17 hladin OHP.
rozhodnutí o léčbě by mělo být založeno na posouzení skutečností a mělo by se řídit individualizovaným přístupem. Léčba není vždy uvedeno, pokud pacient je symptomatický, například, děti s časným nástupem a rychlé progresi ochlupení a ochlupení, rychlý růst, a/nebo kosterní rozvoj, nebo ženy s oligomenorhey, akné, nadměrné ochlupení, neplodnost, nebo kombinace těchto a dalších výše uvedených příznaků. Genetické poradenství se důrazně doporučuje u žen NCCAH, které si přejí otěhotnět, stejně jako genotypizace otce. Celková léčba pacienta navíc zahrnuje zvládnutí pravděpodobných komplikací léčby glukokortikoidy nebo projevů onemocnění souvisejících s metabolismem. A konečně, vzhledem k tomu, že mnoho terapeutické otázky týkající se řádné správy těchto pacientů nebyly dosud objasněn, je velmi důležité pro ošetřujícího lékaře, aby držet krok se všemi vývoj v této oblasti a integrovat nové údaje do své klinické praxe. Další klinické studie v této oblasti jsou jistě nezbytné.
Abych to shrnul, pro NCCAH žena, ideální přístup je šitý na míru, se zabudovanými hladký přechod z její řízení, jakmile ona je odkazoval se z pediatrické do dospělé endokrinolog, spolu s příznakem-orientované léčby, která bude doprovázet ji po celý její život. Vzhledem k mnoha faktorům ovlivňujícím hyperandrogenní systém je vhodné povzbudit pacienty, aby přijali zdravější životní styl zlepšením svých stravovacích návyků, zvýšením cvičení a zaměřením na snížení hmotnosti. Kromě toho je v některých případech psychologická podpora často prospěšná. Navíc specialisté v oborech zapojených do léčby této poruchy, jako jsou dermatologové, gynekologové a psychologové, potřebují důkladné porozumění o řízení podezřelých nebo již diagnostikovaných případů NCCAH. Na závěr mějte na paměti zasvěcený citát amerického astronoma very Coopera Rubina: „věda postupuje nejlépe, když nás pozorování nutí změnit naše předsudky.“
Autor Příspěvky
Všichni autoři uvedených mít podstatné, přímé a intelektuální příspěvek k práci, a to schválil ke zveřejnění.
Prohlášení o střetu zájmů
autoři prohlašují, že výzkum byl proveden bez jakýchkoli obchodních nebo finančních vztahů, které by mohly být vykládány jako potenciální střet zájmů.
1. Speiser PW, bílý PC. Vrozená hyperplazie nadledvin. N Engl J Med. (2003) 349:776–88. doi: 10.1056/NEJMra021561
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
2. Turcu AF, Auchus RJ. Adrenal steroidogenesis and congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. (2015) 44:275–96. doi: 10.1016/j.ecl.2015.02.002
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Speiser PW. Nonclassic adrenal hyperplasia. Rev Endocr Metab Disord. (2009) 10:77–82. doi: 10.1007/s11154-008-9097-x
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Gidlöf S, Falhammar H, Thilén A, von Döbeln U, Ritzén M, Wedell A, et al. Sto let vrozené adrenální hyperplazie ve Švédsku: retrospektivní kohortová studie založená na populaci. Lancet Diabetes Endocrinol. (2013) 1:35–42. doi: 10.1016/S2213-8587(13)70007-X
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
5. Speiser PW, Dupont B, Rubinstein P, Piazza a, Kastelan A, New MI. Vysoká frekvence neklasického deficitu steroidní 21-hydroxylázy. Am J Hum Genet. (1985) 37:650–67.
PubMed Abstract | Google Scholar
6. Wilson RC, Nimkarn S, Dumic M, Obeid J, Azar MR, Azar M, et al. Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Mol Genet Metab. (2007) 90:414–21. doi: 10.1016/j.ymgme.2006.12.005
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Miller WL. Clinical review 54: genetics, diagnosis, and management of 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1994) 78:241–6. doi: 10.1210/jcem.78.2.8106606
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Witchel SF, Azziz R. Nonclassic congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2010) 2010:625105. doi: 10.1186/1687-9856-2010-625105
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Wilson RC, Mercado AB, Cheng KC, nový MI. Nedostatek steroidů 21-hydroxylázy: genotyp nemusí předpovídat fenotyp. J Clin Endocrinol Metab. (1995) 80:2322–9. doi: 10.1210 / jc.80.8.2322
CrossRef Plný Text | Google Scholar
10. Bílý PC. Novorozenecký screening vrozené adrenální hyperplazie. Nat Rev Endocrinol. (2009) 5:490–8. doi: 10.1038 / nrendo.2009.148
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Kohn B, Levine LS, Pollack MS, Pang S, Lorenzen F, Levy D, et al. Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. (1982) 55:817–27. doi: 10.1210/jcem-55-5-817
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Moran C, Azziz R, Carmina E, Dewailly D, Fruzzetti F, Ibañez L, et al. Neklasická adrenální hyperplazie s deficitem 21-hydroxylázy je progresivní porucha: multicentrická studie. Jsem J Porodník Gynecol. (2000) 183:1468–74. doi: 10.1067 / mob.2000.108020
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
13. Azziz R, Hincapie LA, Knochenhauer ES, Dewailly D, Fox L, Boots LR. Screening neklasické adrenální hyperplazie s deficitem 21-hydroxylázy u hyperandrogenních žen: prospektivní studie. Fertil Steril. (1999) 72:915–25. doi: 10.1016/S0015-0282(99)00383-0
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
14. Livadas S, Dracopoulou M, Dastamani A, Sertedaki A, Maniati-Christidi M, Magiakou A-M, et al. Spektrum klinických, hormonálních a molekulárních nálezů u 280 jedinců s neklasickou vrozenou adrenální hyperplazií způsobenou mutacemi genu CYP21A2. Clin Endokrinol (Oxf). (2015) 82:543–9. doi: 10.1111 / cen.12543
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
15. Bidet M, Bellanné-Chantelot C, Galand-Portier M-B, Tardy V, Billaud L, Laborde K, et al. Klinická a molekulární charakterizace kohorty 161 nesouvisí ženy s neklasické kongenitální adrenální hyperplazie v důsledku 21-hydroxylázy deficit a 330 členů rodiny. J Clin Endocrinol Metab. (2009) 94:1570–8. doi: 10.1210 / jc.2008-1582
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
16. Escobar-Morreale HF, Sanchón R, San Millán ml. Prospektivní studie prevalence neklasické vrozené adrenální hyperplazie u žen s hyperandrogenními příznaky a příznaky. J Clin Endocrinol Metab. (2008) 93:527–33. doi: 10.1210 / jc.2007-2053
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
17. Dewailly D, Vantyghem-Haudiquet MC, Sainsard C, Buvat J, Cappoen JP, Ardaens K, et al. Klinické a biologické fenotypy při pozdním nástupu deficitu 21-hydroxylázy. J Clin Endocrinol Metab. (1986) 63:418–23. doi: 10.1210/jcem-63-2-418
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
18. Gönç EN, Ozön ZA, Alikaşifoglu A, Engiz O, Bulum B, Kandemir N. (2011). Je bazální sérum 17-OH progesteron spolehlivým parametrem pro předpovídání neklasické vrozené adrenální hyperplazie u předčasných adrenarche? Turek J. 53:274–80.
PubMed Abstraktní / Google Scholar
19. Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, et al. Vrozená adrenální hyperplazie způsobená nedostatkem steroidní 21-hydroxylázy: směrnice klinické praxe Endokrinní společnosti. J Clin Endocrinol Metab. (2010) 95:4133–60. doi: 10.1210 / jc.2009-2631
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Dacou-Voutetakis C, Dracopoulou M. High incidence of molecular defects of the CYP21 gene in patients with premature adrenarche. J Clin Endocrinol Metab. (1999) 84:1570–4. doi: 10.1210/jc.84.5.1570
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Ambroziak U, Kepczynska-Události A Курылович, Małunowicz EM, Wójcicka, Мискевич P, et al. The diagnosis of nonclassic kongenitální adrenální hyperplazie due to 21-hydroxylase deficiency, based on sérum basal or post-ACTH stimulation 17-hydroxyprogesterone, can lead to false-positive diagnosis. Clin Endocrinol (Oxf). (2016) 84:23–9. doi: 10.1111 / cena.12935
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Ambroziak U, Kepczynska-Události A Курылович, Wysłouch-Cieszyńska A, Małunowicz EM, Bartoszewicz, et al. LC-MS/MS improves screening towards 21-hydroxylase deficiency. Gynecol Endokrinol. (2015) 31:296–300. doi: 10.3109/09513590.2014.994599
PubMed Abstract | CrossRef Full Text | Google Scholar
23. BBC, CC, Goldman mm, Zhang k, Clark-N, Writz RE Welt CK. Současné měření třinácti steroidních hormonů u žen se syndromem polycystických vaječníků a kontrolní ženy pomocí kapalinové chromatografie-tandemové hmotnostní spektrometrie. PLoS Jedna. (2014) 9: e93805. doi: 10.1371 / deník.pone.0093805
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
24. Ray JA, Kushnir MM, Yost RA, Rockwood AL, Wayne Meikle a. zvýšení výkonu při měření 5 endogenních steroidů pomocí LC-MS / MS v kombinaci se spektrometrií diferenciální iontové mobility. Clin Chim Acta. (2015) 438:330–6. doi: 10.1016 / j. cca.2014.07.036
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
25. Stoupa A, González-Briceño L, Pinto G, Samara-Boustani D, Thalassinos C, Flechtner I, et al. Nedostatečná odpověď kortizolu na test tetracosactide (Synacthen®) u neklasické vrozené adrenální hyperplazie: výjimka z pravidla? Horm Res Paediatr. (2015) 83:262–7. doi: 10.1159/000369901
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. (1992) 90:584–95. doi: 10.1172/JCI115897
PubMed Abstract | CrossRef Full Text | Google Scholar
27. Temeck JW, Pang SY, Nelson C, nový MI. Genetické defekty steroidogeneze v předčasném pubarche. J Clin Endocrinol Metab. (1987) 64:609–17. doi: 10.1210/jcem-64-3-609
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
28. Voutilainen R, Jääskeläinen J. předčasné adrenarche: etiologie, klinické nálezy a důsledky. J Steroid Biochem Mol Biol. (2015) 145:226–36. doi: 10.1016 / j. jsbmb.2014.06.004
PubMed Abstract | CrossRef Full Text | Google Scholar
29. Oberfield SE, Sopher AB, Gerken AT. Approach to the girl with early onset of pubic hair. J Clin Endocrinol Metab. (2011) 96:1610–22. doi: 10.1210/jc.2011-0225
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Janus D, Wojcik M, Malunowicz E, Starzyk JB. Případ opakujících se labiálních adhezí u 15měsíčního dítěte s asymptomatickou neklasickou vrozenou adrenální hyperplazií v důsledku nedostatku 21-hydroxylázy. J Pediatr Endocrinol Metab. (2012) 25:1017–21. doi: 10.1515/jpem-2012-0190
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
31. Gopal-Kothandapani JS, Petkar A, O ‚ Shea E, Banerjee i. perianální vlasy jako neobvyklá prezentace neklasické vrozené adrenální hyperplazie. BMJ věc Rep. (2013) 2013: bcr2013010123. doi: 10.1136/bcr-2013-010123
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Falhammar H, Nordenström A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine. (2015) 50:32–50. doi: 10.1007/s12020-015-0656-0
PubMed Abstract | CrossRef Full Text | Google Scholar
33. Chrousos GP, Loriaux DL, Mann DL, Cutler GB. Late-onset 21-hydroxylase deficiency mimicking idiopathic hirsutism or polycystic ovarian disease. Ann Intern Med. (1982) 96:143–8. doi: 10.7326/0003-4819-96-2-143
CrossRef Full Text | Google Scholar
34. Witchel SF. Non-classic congenital adrenal hyperplasia. Steroids. (2013) 78:747–50. doi: 10.1016/j.steroids.2013.04.010
PubMed Abstract | CrossRef Full Text | Google Scholar
35. Carmina E, Rosato F, Jannì A, Rizzo M, Longo RA. Extensive clinical experience: relative prevalence of different androgen excess disorders in 950 women referred because of clinical hyperandrogenism. J Clin Endocrinol Metab. (2006) 91:2–6. doi: 10.1210/jc.2005-1457
PubMed Abstract | CrossRef Full Text | Google Scholar
36. Moran C, Azziz R, Weintrob N, Witchel SF, Rohmer V, Dewailly D, et al. Reprodukční výsledek žen s neklasickou adrenální hyperplazií s deficitem 21-hydroxylázy. J Clin Endocrinol Metab. (2006) 91:3451–6. doi: 10.1210 / jc.2006-0062
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
37. Levin JH, Carmina E, Lobo RA. Je nevhodná sekrece gonadotropinu u pacientů se syndromem polycystických vaječníků podobná sekreci pacientů s vrozenou adrenální hyperplazií u dospělých? Fertil Steril. (1991) 56:635–40. doi: 10.1016/S0015-0282(16)54592-0
PubMed Abstract | CrossRef Full Text | Google Scholar
38. Carmina E, Lobo RA. Ovarian suppression reduces clinical and endocrine expression of late-onset congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Fertil Steril. (1994) 62:738–43. doi: 10.1016/S0015-0282(16)56998-2
PubMed Abstract | CrossRef Full Text | Google Scholar
39. Ludwig e. klasifikace typů androgenetické alopecie (běžné plešatosti) vyskytující se u ženského pohlaví. Br J Dermatol. (1977) 97:247–54. doi: 10.1111 / j. 1365-2133.1977.tb15179.x
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
40. Trakakis E, Papadavid E, Dalamaga M, Koumaki D, Stavrianeas N, Rigopoulos D, et al. Prevalence neklasické vrozené adrenální hyperplazie v důsledku nedostatku 21-hydroxylázy u řeckých žen s akné: průřezová studie založená na nemocnici. J Eur Acad Dermatol Venereol. (2013) 27:1448–51. doi: 10.1111 / j. 1468-3083. 2012. 04613.x
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
41. Arlt W, Willis DS, Wild SH, Krone N, Doherty EJ, Hahner S, et al. Zdravotní stav dospělých s vrozenou adrenální hyperplazií: kohortová studie 203 pacientů. J Clin Endocrinol Metab. (2010) 95:5110–21. doi: 10.1210 / jc.2010-0917
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
42. Saygili F, Oge, Yilmaz C. Hyperinzulinémií a necitlivosti u žen s neklasické kongenitální adrenální hyperplazie v důsledku 21-hydroxylázy deficit: vztah mezi sérové hladiny leptinu a chronické hyperinzulinémie. Horm Res. (2005) 63: 270-4. doi: 10.1159/000086363
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Speiser PW, Serrat J, New MI, Gertner JM. Insulin insensitivity in adrenal hyperplasia due to nonclassical steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. (1992) 75:1421–4. doi: 10.1210/jc.75.6.1421
PubMed Abstract | CrossRef Full Text | Google Scholar
44. Han TS, Stimson RH, Rees DA, Krone N, Willis DS, Conway GS, et al. Režim léčby glukokortikoidy a zdravotní výsledky u dospělých s vrozenou adrenální hyperplazií. Klinický Endokrinol. (2013) 78:197–203. doi: 10.1111 / cen.12045
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
45. Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. použití perorálních kortikosteroidů a riziko zlomenin. J Bone Miner Res. (2000) 15: 993-1000. doi: 10.1359 / jbmr.2000.15.6.993
PubMed Abstract | CrossRef Full Text | Google Scholar
46. Nebesio TD, Renbarger JL, Nabhan ZM, Ross SE, Slaven JE, Li L, et al. Differential effects of hydrocortisone, prednisone, and dexamethasone on hormonal and pharmacokinetic profiles: a pilot study in children with congenital adrenal hyperplasia. Int J Pediatr Endocrinol. (2016) 2016:17. doi: 10.1186/s13633-016-0035-5
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
47. Weintrob N, Dickerman Z, Sprecher E, Galatzer, Pertzelan A. Non-klasický 21-hydroxylázy deficit v kojeneckém věku a dětství: vliv doby zahájení léčby, v pubertě a konečné výšky. Eur J Endokrinol. (1997) 136:188–95. doi: 10.1530 / eje.0.1360188
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Bonfig W, Bechtold S, Schmidt H, Knorr D, Schwarz HP. Reduced final height outcome in congenital adrenal hyperplasia under prednisone treatment: deceleration of growth velocity during puberty. J Clin Endocrinol Metab. (2007) 92:1635–9. doi: 10.1210/jc.2006-2109
PubMed Abstract | CrossRef Full Text | Google Scholar
49. Dörr HG. Růst u pacientů s klasickou vrozenou adrenální hyperplazií v důsledku nedostatku 21-hydroxylázy. Horm Res. (2007) 68 (Suppl 5): 93-9. doi: 10.1159/000110587
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
50. Hoepffner W, Kaufhold, Willgerodt H, Keller, E. Pacienti s klasickou kongenitální adrenální hyperplazie v důsledku 21-hydroxylázy deficit může dosáhnout své cílové výšky: Lipsko zkušenosti. Horm Res. (2008) 70: 42-50. doi: 10.1159/000129677
PubMed Abstract | CrossRef Full Text | Google Scholar
51. van der Kaay D, van den Akker E. Ultralow-dose dexamethasone to preserve endogenous cortisol stress response in nonclassical congenital adrenal hyperplasia: a new promising treatment. Int J Endocrinol Metab. (2014) 12:e14657. doi: 10.5812/ijem.14657
CrossRef Full Text | Google Scholar
52. Lin-Su K, Vogiatzi MG, Marshall I, Harbison MD, Macapagal MC, Betensky B, et al. Treatment with growth hormone and luteinizing hormone releasing hormone analog improves final adult height in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. (2005) 90:3318–25. doi: 10.1210/jc.2004-2128
PubMed Abstract | CrossRef Full Text | Google Scholar
53. McCann-Crosby B, Chen M-J, Lyons SK, Lin Y, Axelrad M, Dietrich JE, et al. Nonclassical congenital adrenal hyperplasia: cíle léčby a přechodu. Pediatr Endocrinol Rev. (2014) 12: 224-38.
PubMed Abstraktní | Google Scholar
54. Ogilvie CM, Crouch NS, Rumsby G, Creighton SM, Liao L-M, Conway GS. Vrozená adrenální hyperplazie u dospělých: přehled lékařských, chirurgických a psychologických problémů. Klinický Endokrinol. (2006) 64:2–11. doi: 10.1111 / j. 1365-2265. 2005. 02410.x
PubMed Abstract | CrossRef Full Text | Google Scholar
55. Mah PM, Jenkins RC, Rostami-Hodjegan A, Newell-Price J, Doane A, Ibbotson V, et al. Weight-related dosing, timing and monitoring hydrocortisone replacement therapy in patients with adrenal insufficiency. Clin Endocrinol. (2004) 61:367–75. doi: 10.1111/j.1365-2265.2004.02106.x
PubMed Abstract | CrossRef Full Text | Google Scholar
56. Diaz A, Laufer Mr závěr LLAA z PC na A, péče AC O O a GC na AH. Menstruace u dívek a dospívajících: použití menstruačního cyklu jako vitálního znamení. Pediatrie. (2006) 118:2245–50. doi: 10.1542 / peds.2006-2481
CrossRef Plný Text | Google Scholar
57. Merke DP, Poppas DP. Léčba adolescentů s vrozenou adrenální hyperplazií. lancet Diabetes Endocrinol. (2013) 1:341–52. doi: 10.1016/S2213-8587(13)70138-4
PubMed Abstract | CrossRef Full Text | Google Scholar
58. Verma S, Green-Golan L, VanRyzin C, Drinkard B, Mehta SP, Weise M, et al. Adrenomedullary function in patients with nonclassic congenital adrenal hyperplasia. Horm Metab Res. (2010) 42:607–12. doi: 10.1055/s-0030-1253385
PubMed Abstract | CrossRef Full Text | Google Scholar
59. New MI. Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2006) 91:4205–14. doi: 10.1210/jc.2006-1645
PubMed Abstract | CrossRef Full Text | Google Scholar
60. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2018) 103:4043–88. doi: 10.1210/jc.2018-01865
PubMed Abstract | CrossRef Full Text | Google Scholar
61. Kruse B, Riepe FG, Krone N, Bosinski HAG, Kloehn S, Partsch CJ, et al. Congenital adrenal hyperplasia – how to improve the transition from adolescence to adult life. Exp Clin Endocrinol Diabetes. (2004) 112:343–55. doi: 10.1055/s-2004-821013
PubMed Abstract | CrossRef Full Text | Google Scholar
62. Gastaud F, Bouvattier C, Duranteau L, Brauner R, Thibaud E, Kutten F, et al. Zhoršené sexuální a reprodukční výsledky u žen s klasickými formami vrozené adrenální hyperplazie. J Clin Endocrinol Metab. (2007) 92:1391–6. doi: 10.1210 / jc.2006-1757
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
63. Mercè Fernández-Balsells M, Muthusamy K, Smushkin G, Lampropulos JC, Elamin MB, Abu Elnour NE, et al. Prenatální dexamethason použít pro prevenci virilizace u těhotenství s rizikem klasické kongenitální adrenální hyperplazie, protože 21-hydroxylázy (CYP21A2) nedostatek: systematický přehled a meta-analýzy. Klinický Endokrinol. (2010) 73:436–44. doi: 10.1111 / j. 1365-2265. 2010. 03826.x
CrossRef Plný Text | Google Scholar
64. Bidet M, Bellanné-Chantelot C, Galand-Portier MB, Golmard JL, Tardy V, Morel Y, et al. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. (2010) 95:1182–90. doi: 10.1210/jc.2009-1383
PubMed Abstract | CrossRef Full Text | Google Scholar
65. Birnbaum MD, Rose LI. Late onset adrenocortical hydroxylase deficiencies associated with menstrual dysfunction. Obstet Gynecol. (1984) 63:445–51.
PubMed Abstract | Google Scholar
66. Cuhaci N, Aydin C, Yesilyurt A, Pinarli FA, Ersoy R, Cakir B. Nonclassical congenital adrenal hyperplasia and pregnancy. Case Rep Endocrinol. (2015) 2015:296924. doi: 10.1155/2015/296924
PubMed Abstract | CrossRef Full Text | Google Scholar
67. Deneux C, Tardy V, Anne D, Mornet E, Billaud L, Charron D, et al. Korelace fenotyp-genotyp u 56 žen s neklasickou vrozenou adrenální hyperplazií v důsledku nedostatku 21-hydroxylázy. J Clin Endocrinol Metab. (2001) 86:207–13. doi: 10.1210 / jcem.86.1.7131
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
68. Simonetti L, Bruque CD, Fernández CS, Benavides-Mori B, Delea M, Kolomenski JE, et al. Aktualizace mutace CYP21A2: komplexní analýza databází a publikovaných genetických variant. Hum Mutat. (2018) 39:5–22. doi: 10.1002 / humu.23351
PubMed Abstract | CrossRef Full Text | Google Scholar
69. OMIM Entry – * 613815 – CYTOCHROME P450 FAMILY 21 SUBFAMILY A POLYPEPTIDE 2; CYP21A2.
Google Scholar
70. Miller WL, Witchel SF. Prenatal treatment of congenital adrenal hyperplasia: risks outweigh benefits. Am J Obstet Gynecol. (2013) 208:354–9. doi: 10.1016/j.ajog.2012.10.885
PubMed Abstract | CrossRef Full Text | Google Scholar
71. Lekarev O, New MI. Adrenal disease in pregnancy. Best Pract Res Clin Endocrinol Metab. (2011) 25:959–73. doi: 10.1016/j.beem.2011.08.004
PubMed Abstract | CrossRef Full Text | Google Scholar
72. New MI, Abraham M, Yuen T, Lekarev O. An update on prenatal diagnosis and treatment of congenital adrenal hyperplasia. Semin Reprod Med. (2012) 30:396–9. doi: 10.1055/s-0032-1324723
PubMed Abstract | CrossRef Full Text | Google Scholar
73. Kazmi D, Bailey J, Yau M, Abu-Amer W, Kumar A, Low M, et al. New developments in prenatal diagnosis of congenital adrenal hyperplasia. J Steroid Biochem Mol Biol. (2017) 165:121–3. doi: 10.1016/j.jsbmb.2016.06.016
PubMed Abstract | CrossRef Full Text | Google Scholar
74. Clayton PE, Miller WL, Oberfield SE, Ritzén EM, SIPPELL WG, Speiser PW, et al. Konsensuální prohlášení o nedostatku 21-hydroxylázy od Evropské společnosti pro pediatrickou endokrinologii a lawson wilkins pediatric endocrine society. Horm Res. (2002) 58: 188-95. doi: 10.1159/000065490
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
75. Nandagopal R, Sinaii N, Avila NA, Van Ryzin C, Chen W, Finkielstain GP, et al. Fenotypové profilování rodičů s kryptickou neklasickou vrozenou adrenální hyperplazií: nálezy ve 145 nesouvisejících rodinách. Eur J Endokrinol. (2011) 164:977–84. doi: 10.1530/EJE-11-0019
PubMed Abstraktní | CrossRef Plný Text | Google Scholar
76. Takasu N, Nakachi K, Higa H. Rozvoje gravesovy hypertyreózy způsobila adrenální krizi u pacienta s dříve nerozpoznané non-classical 21-hydroxylázy deficit. Internistka. (2010) 49:1395–400. doi: 10.2169 / internalmedicine.49.3573
PubMed Abstract | CrossRef Full Text | Google Scholar
77. Meyer-Bahlburg HFL, Dolezal C, Baker SW, Carlson AD, Obeid JS, New MI. Prenatal androgenization affects gender-related behavior but not gender identity in 5-12-year-old girls with congenital adrenal hyperplasia. Arch Sex Behav. (2004) 33:97–104. doi: 10.1023/B:ASEB.0000014324.25718.51
CrossRef Full Text | Google Scholar
78. Meyer-Bahlburg HFL, Doležal C, Baker SW, New MI. Sexuální orientace u žen s klasickou nebo neklasickou vrozenou adrenální hyperplazií jako funkce stupně prenatálního nadbytku androgenu. Arch Sex Chovat. (2008) 37:85–99. doi: 10.1007/s10508-007-9265-1
PubMed Abstraktní | CrossRef Plný Text | Google Scholar